


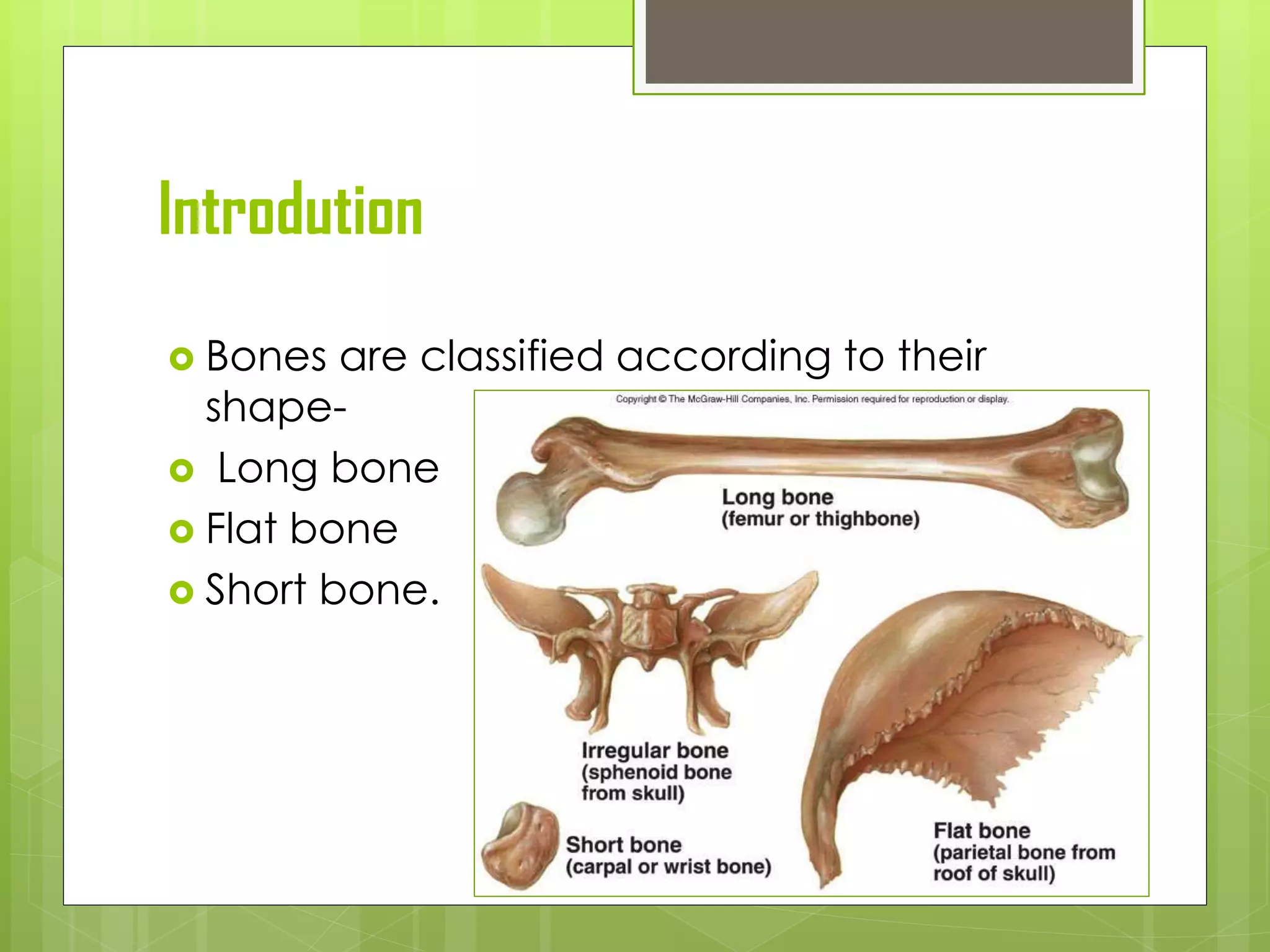

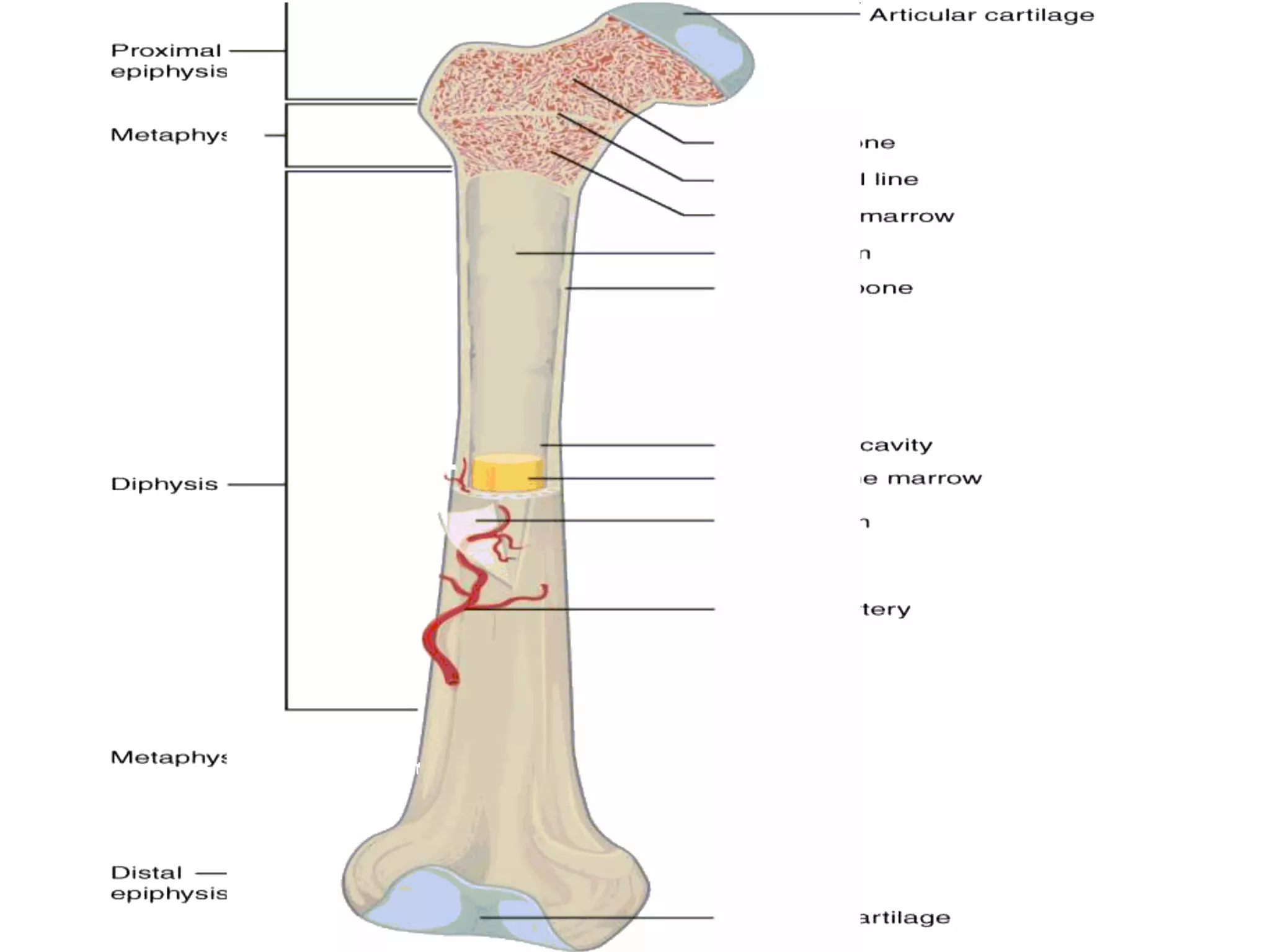
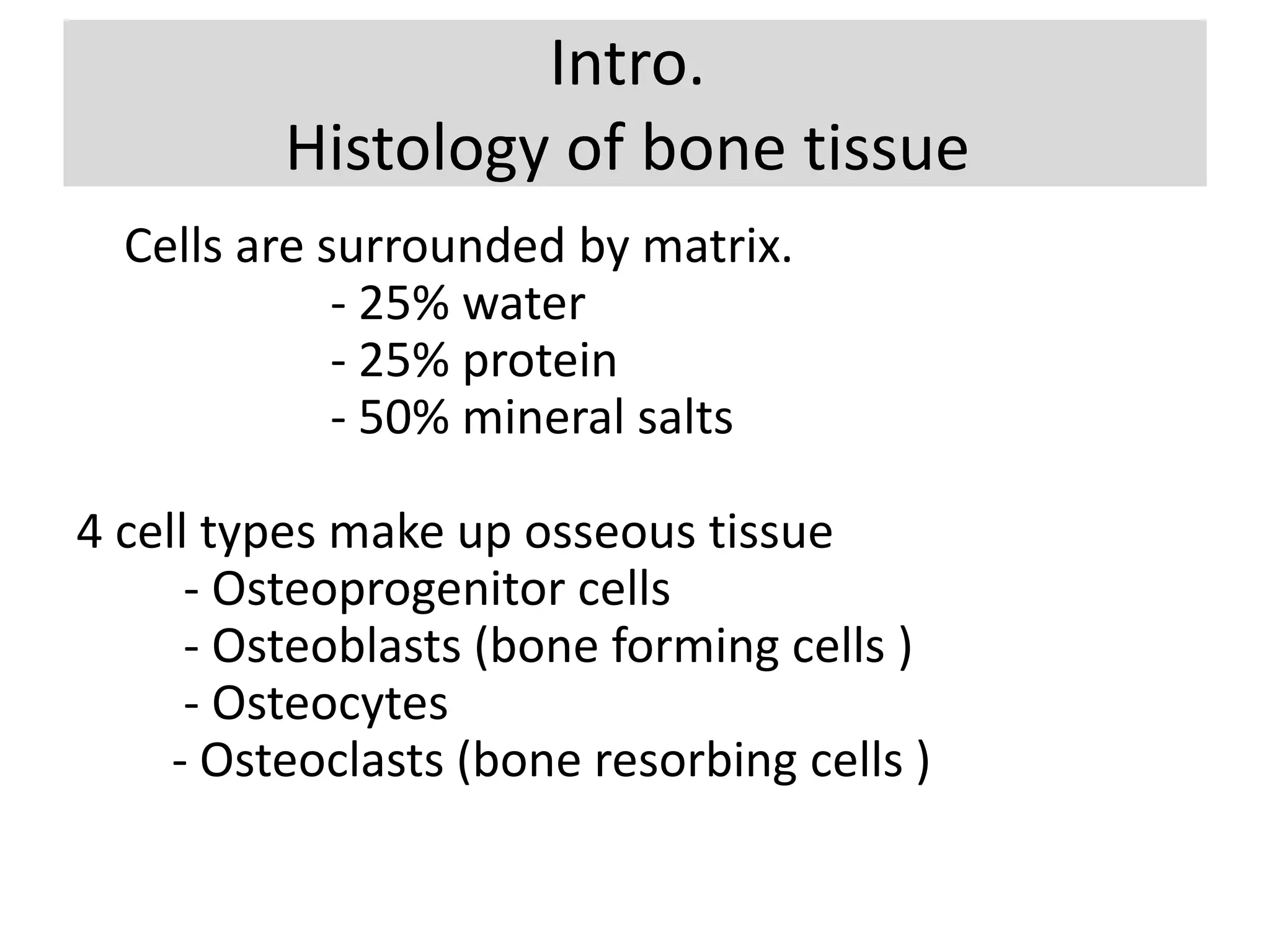
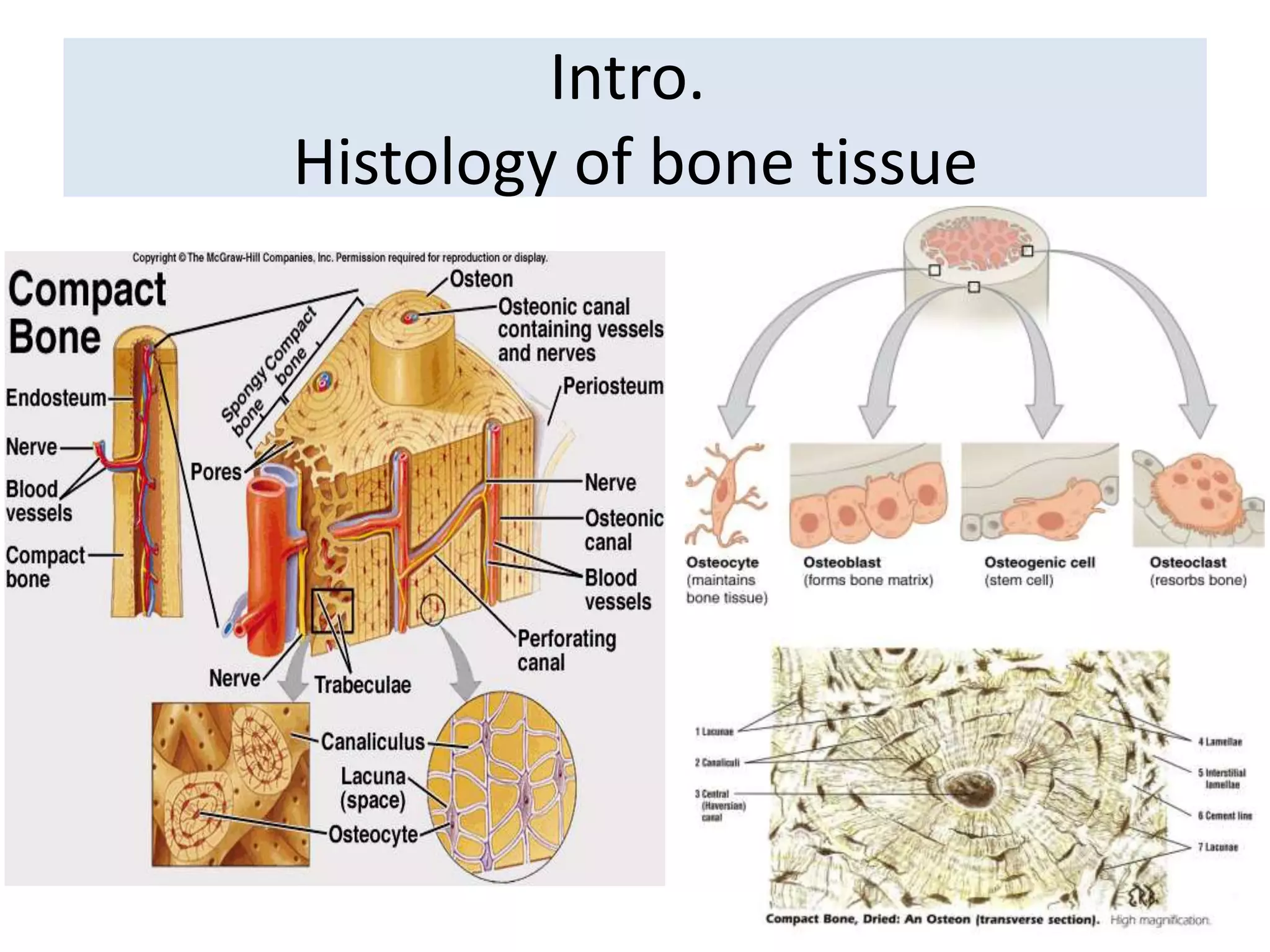
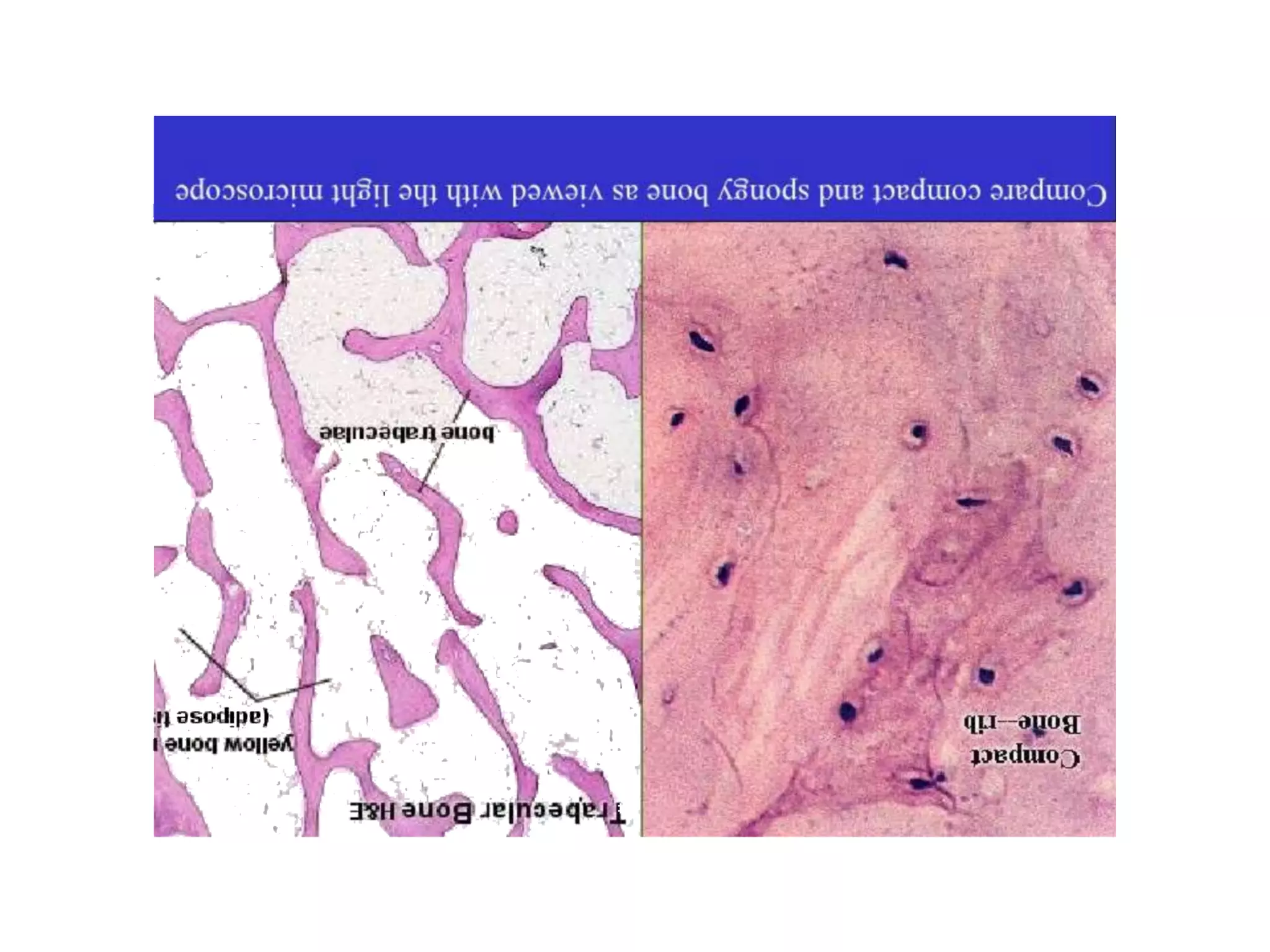


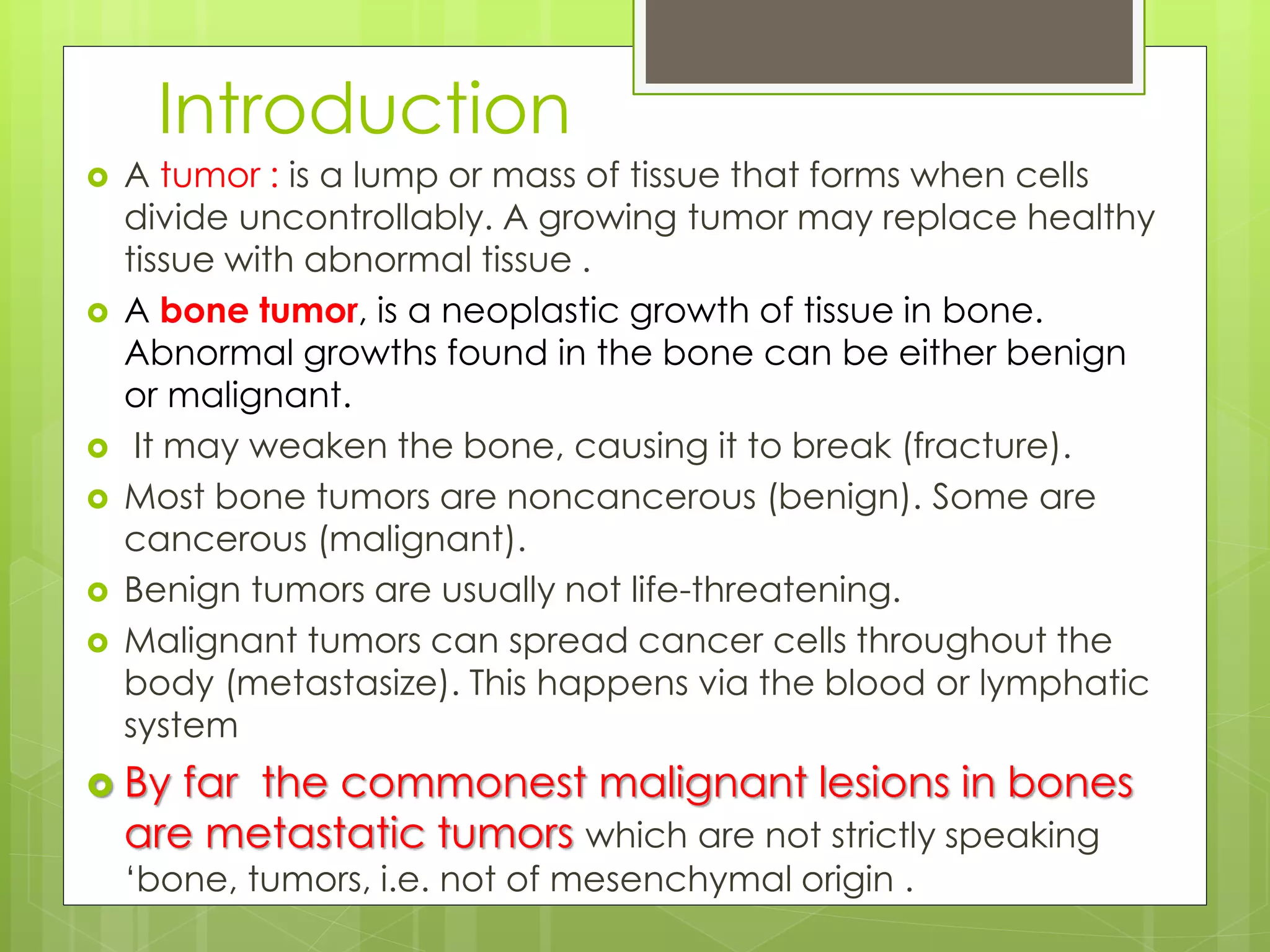









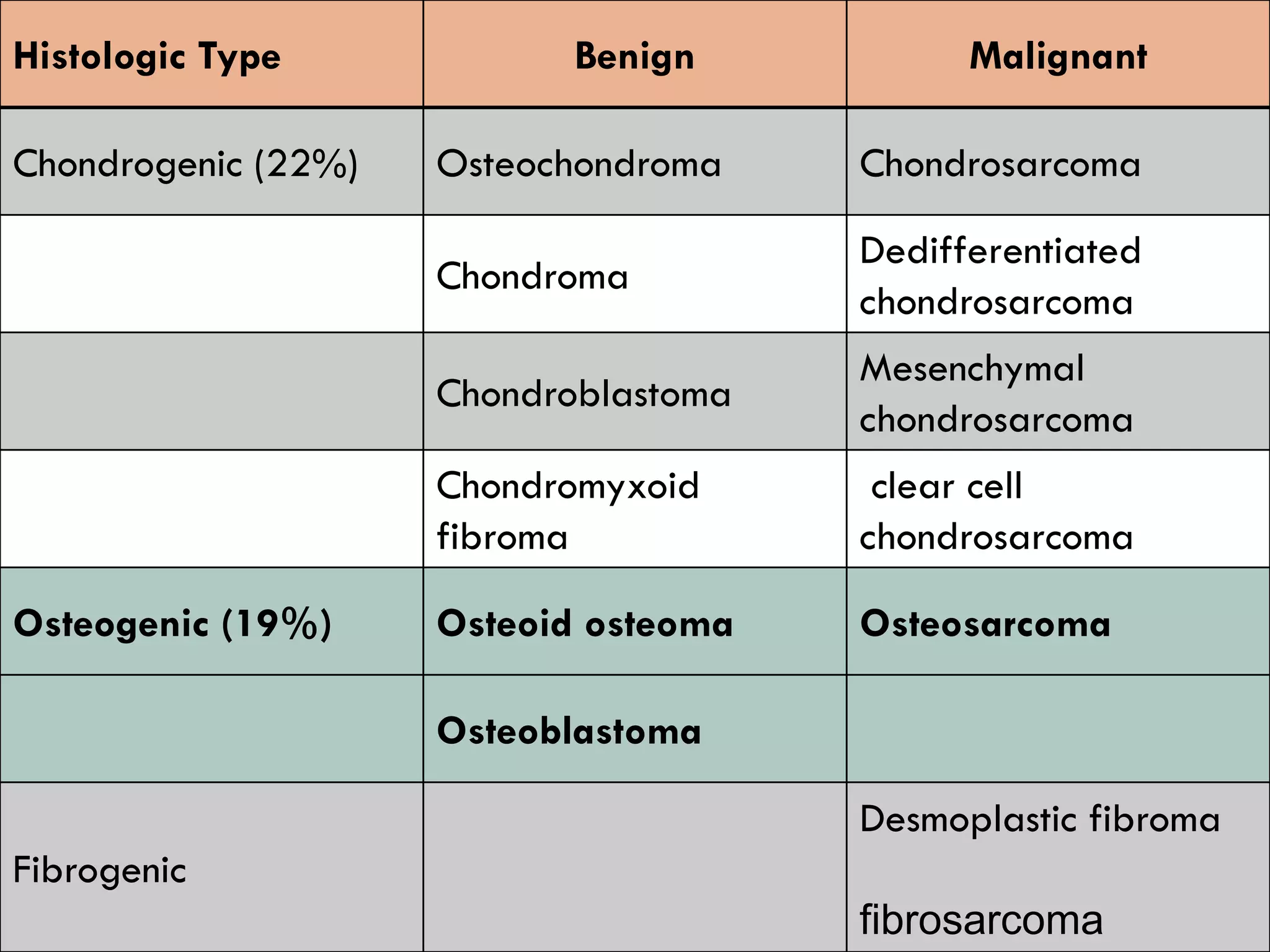
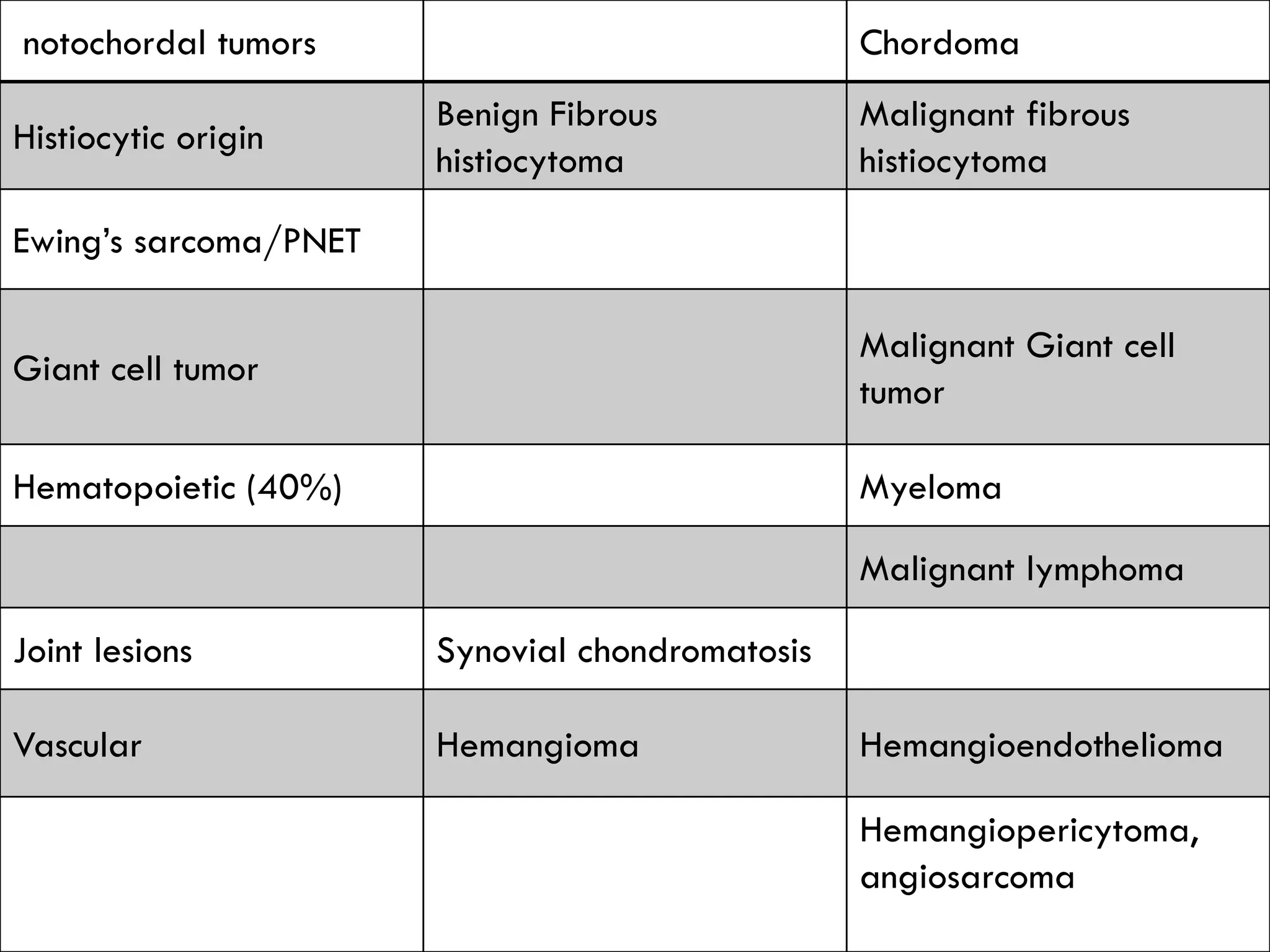
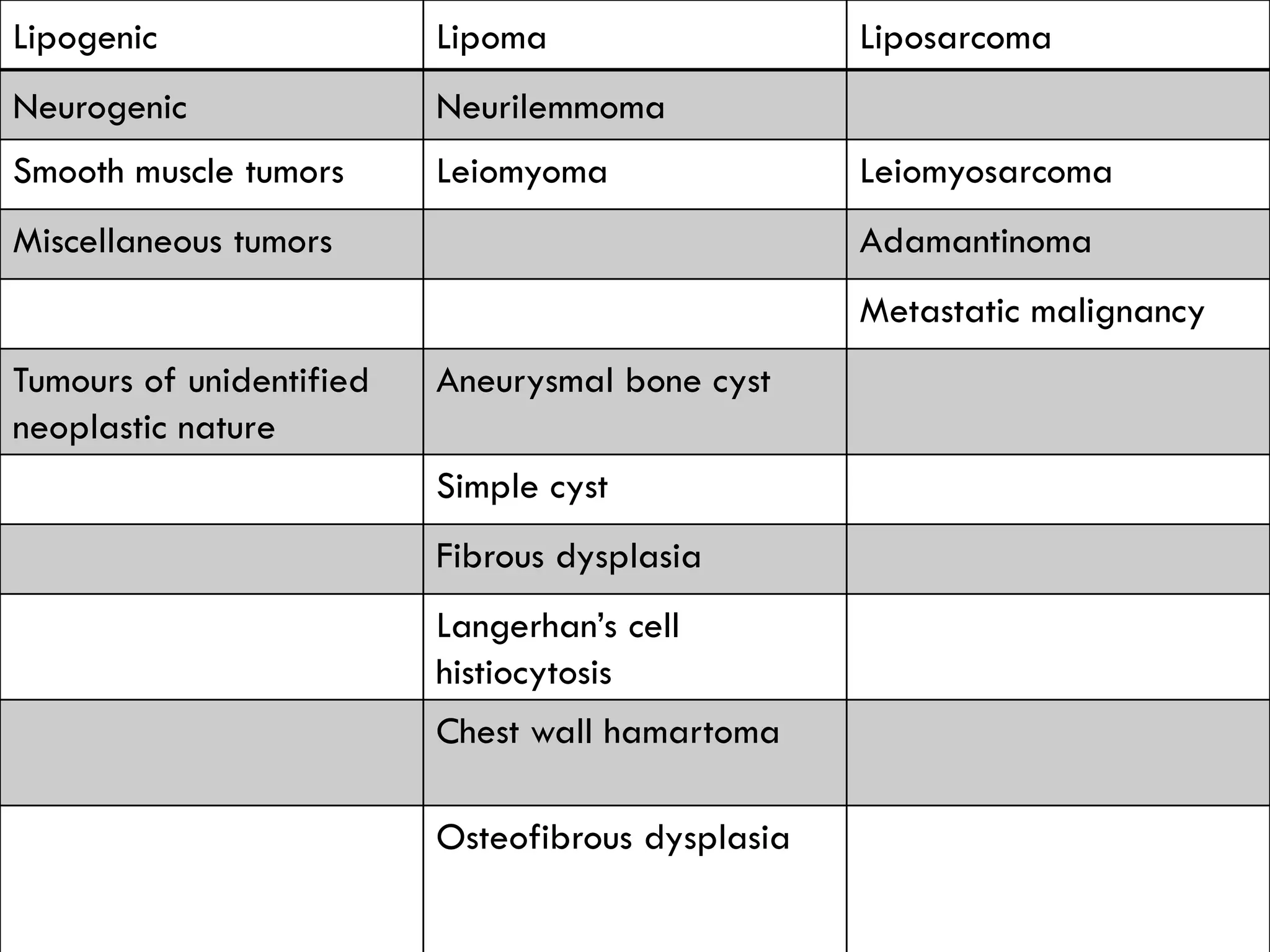
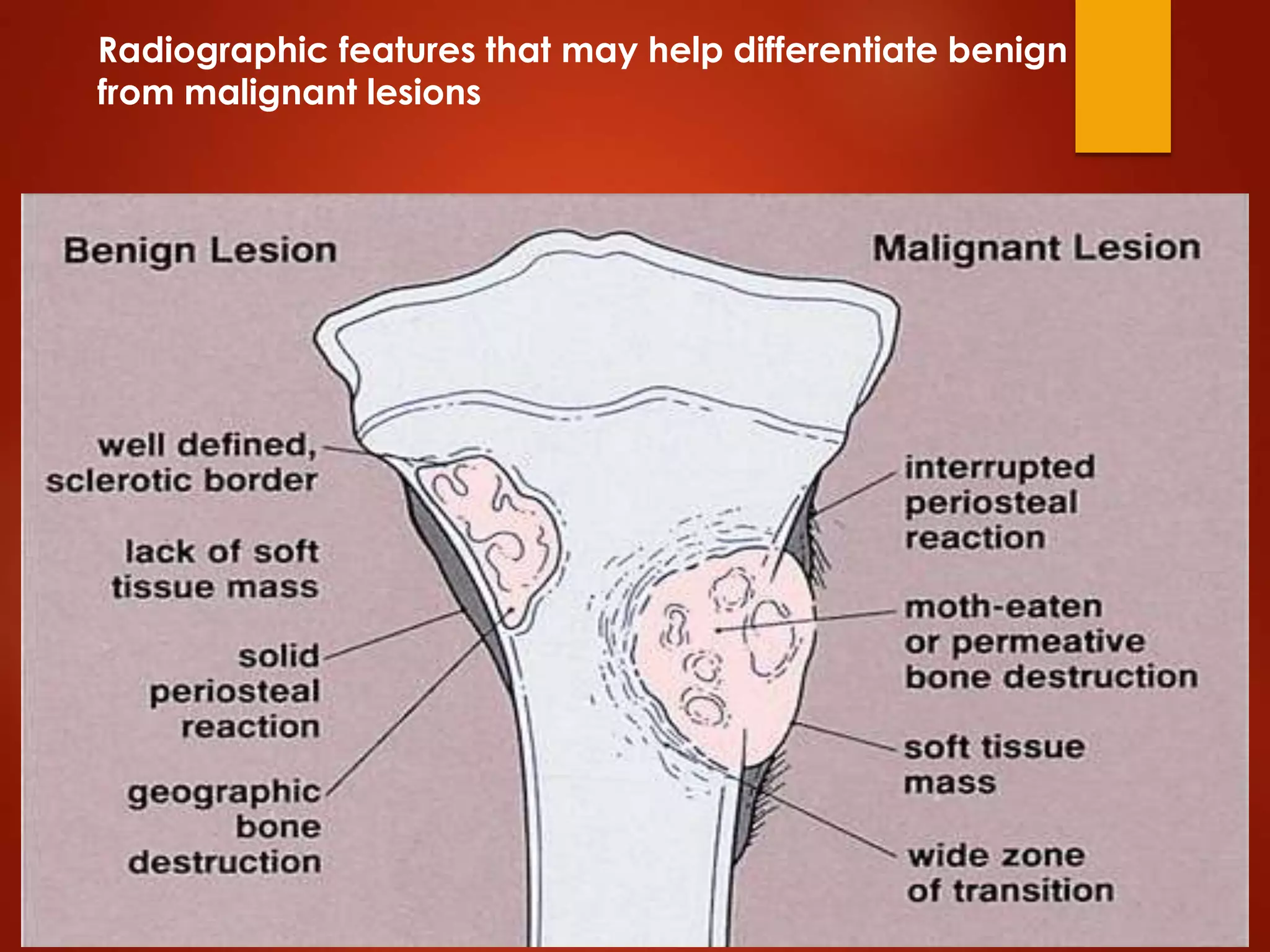


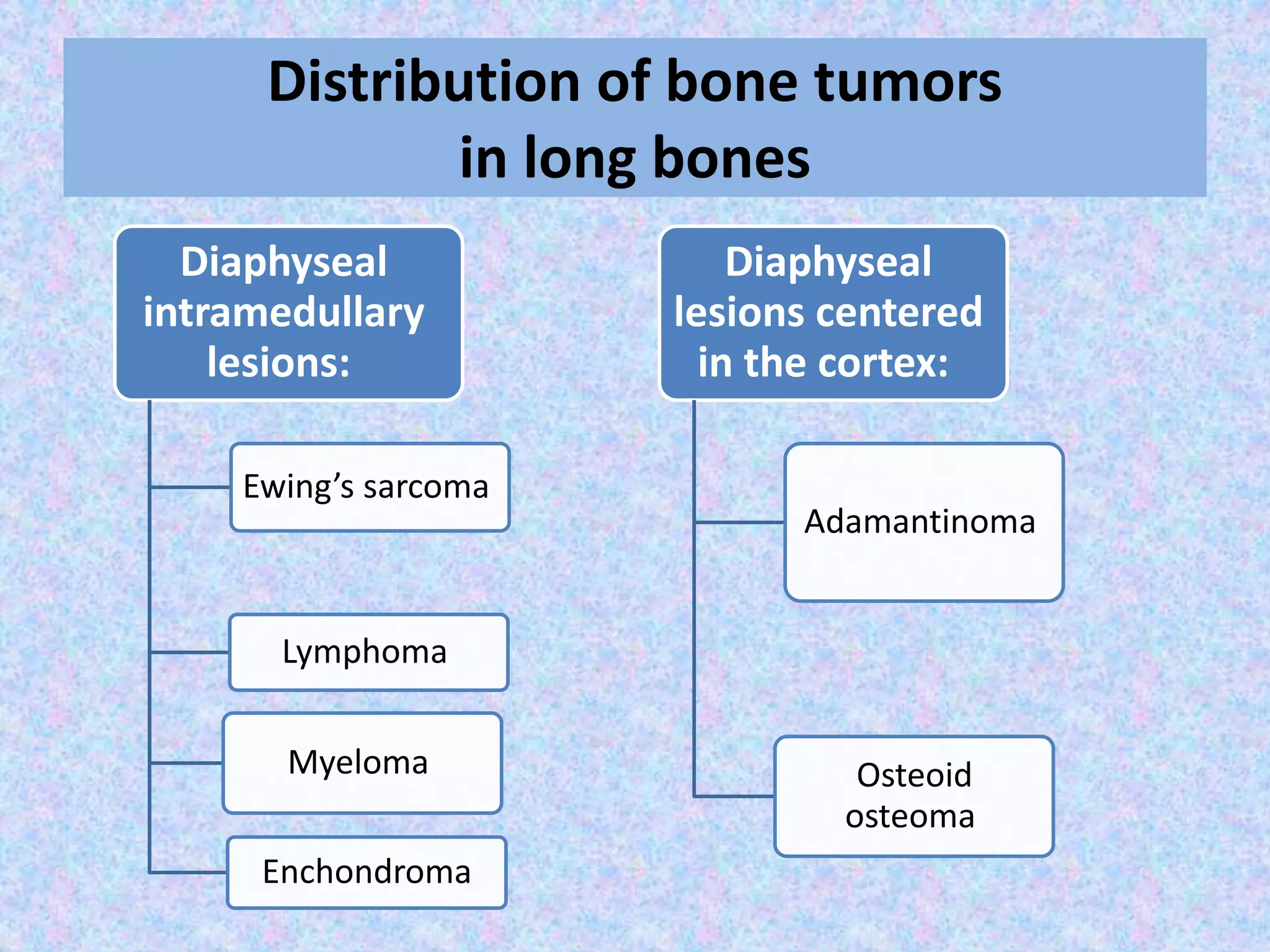




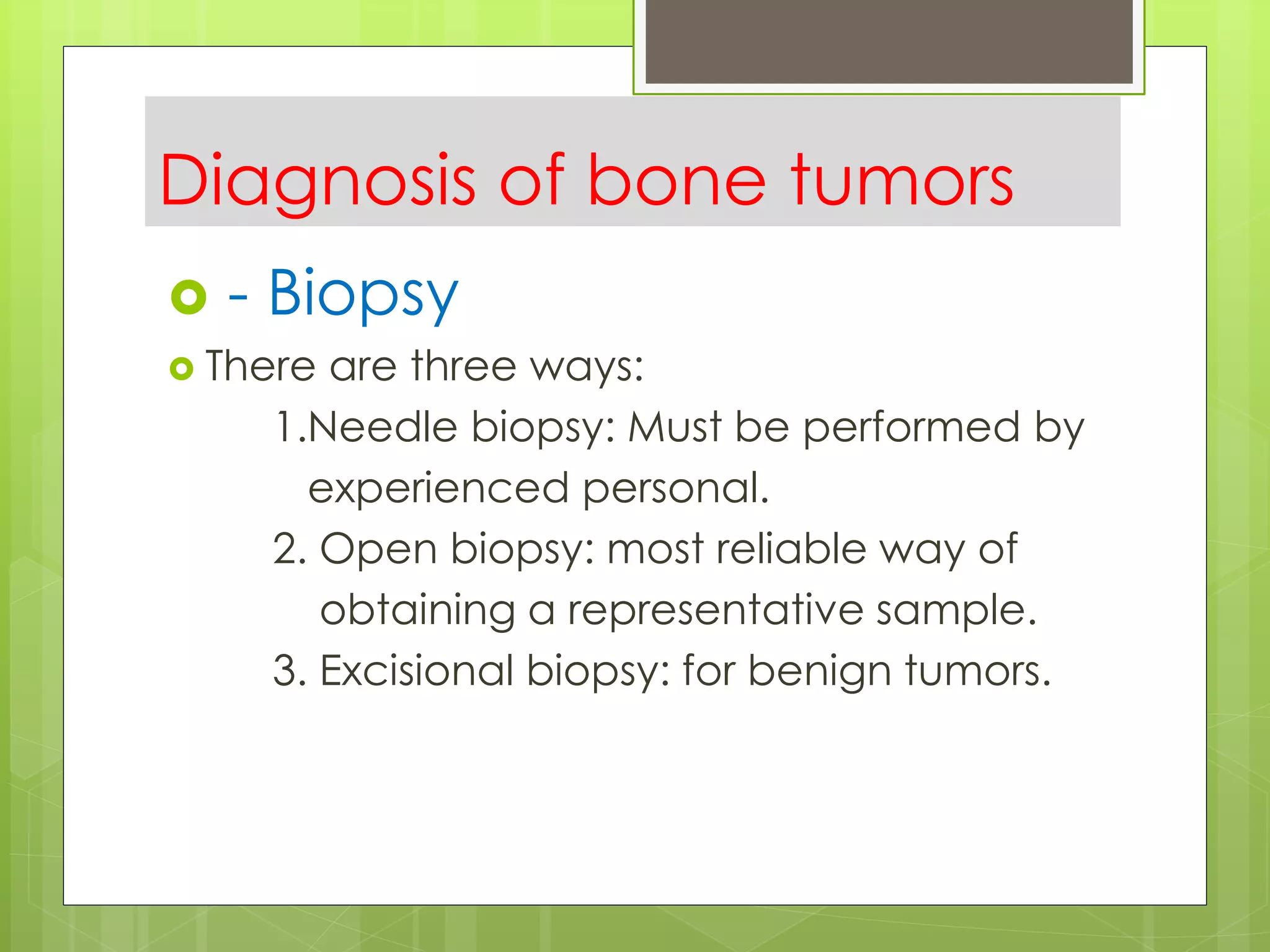










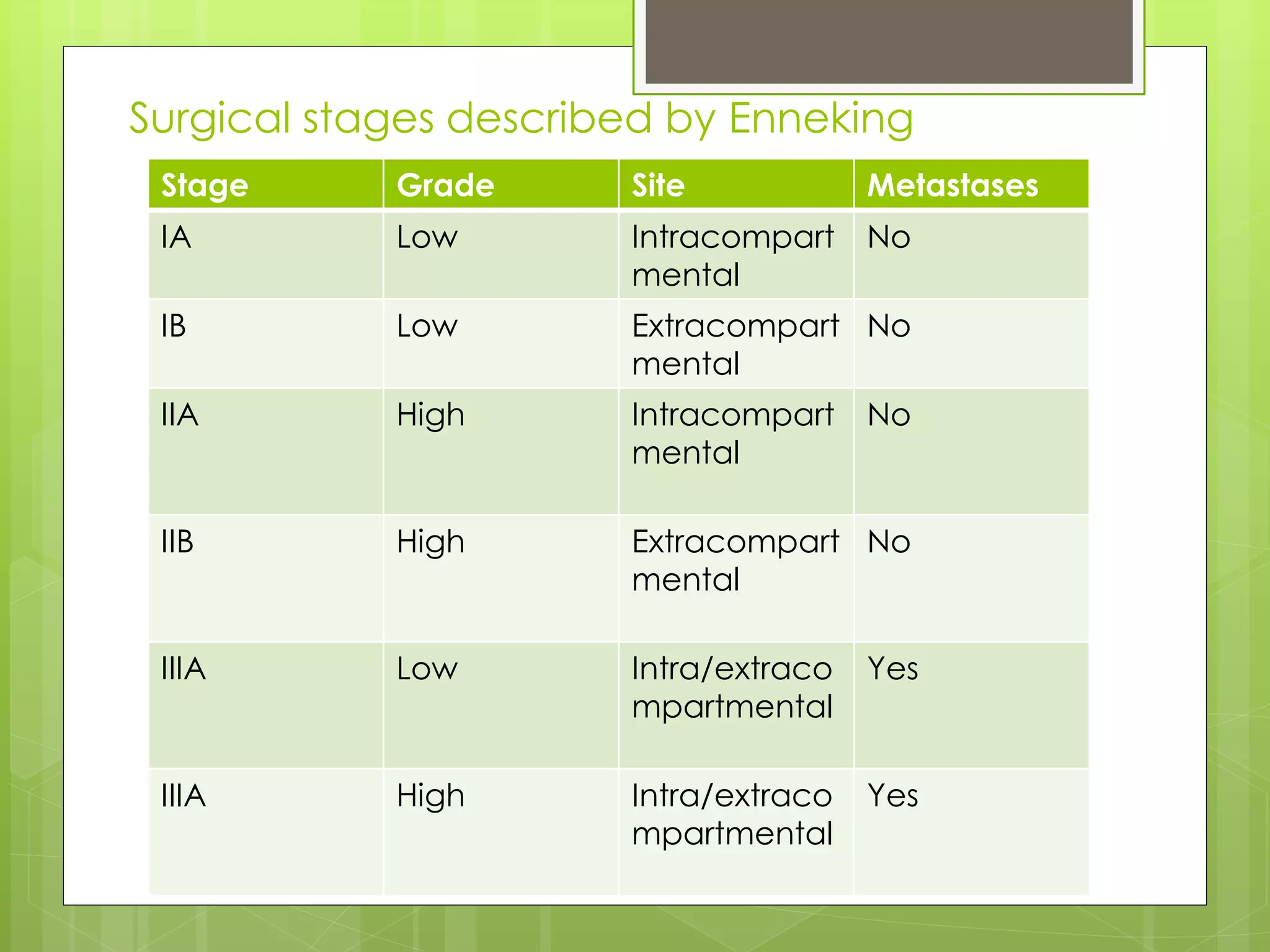

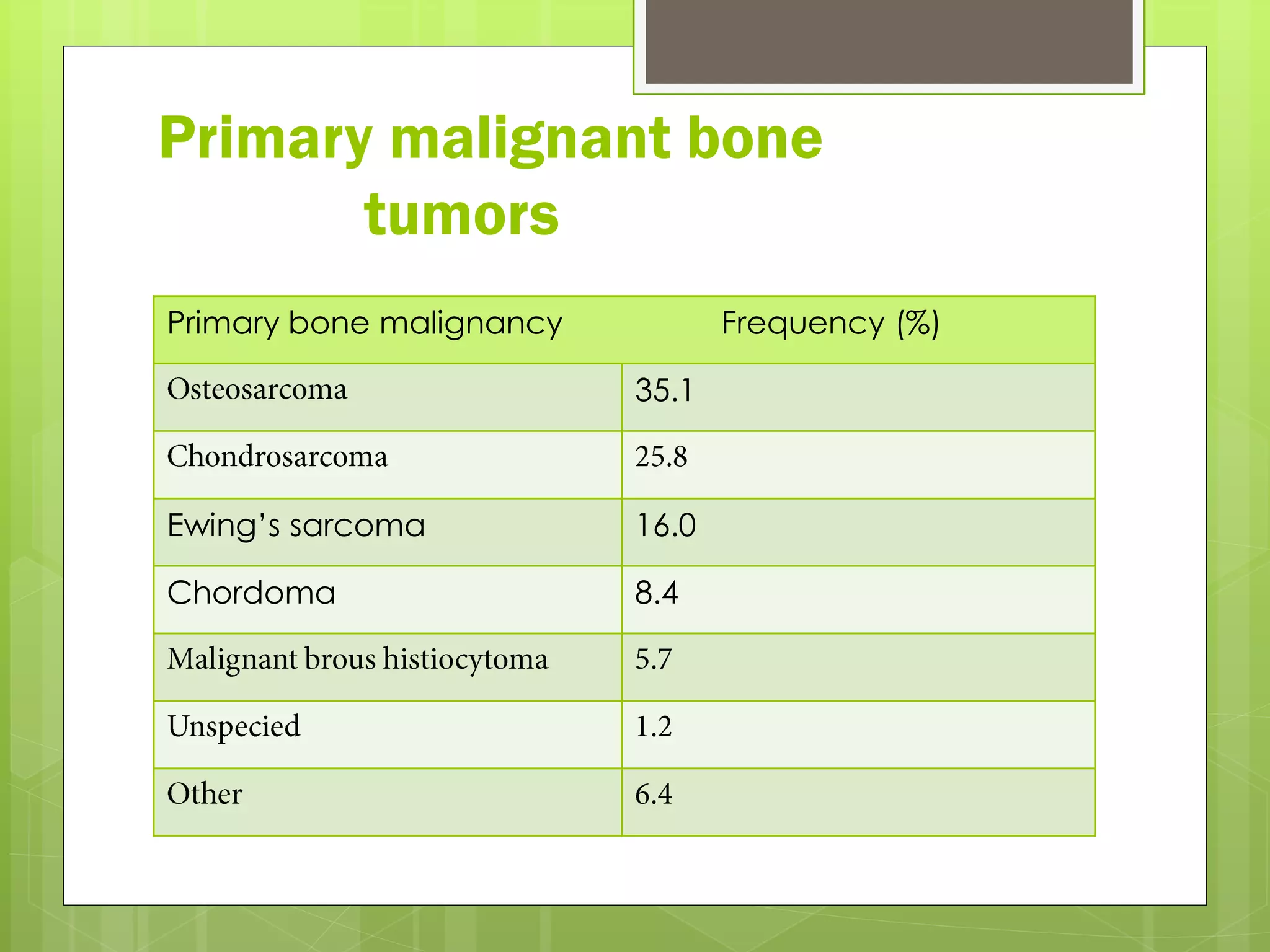

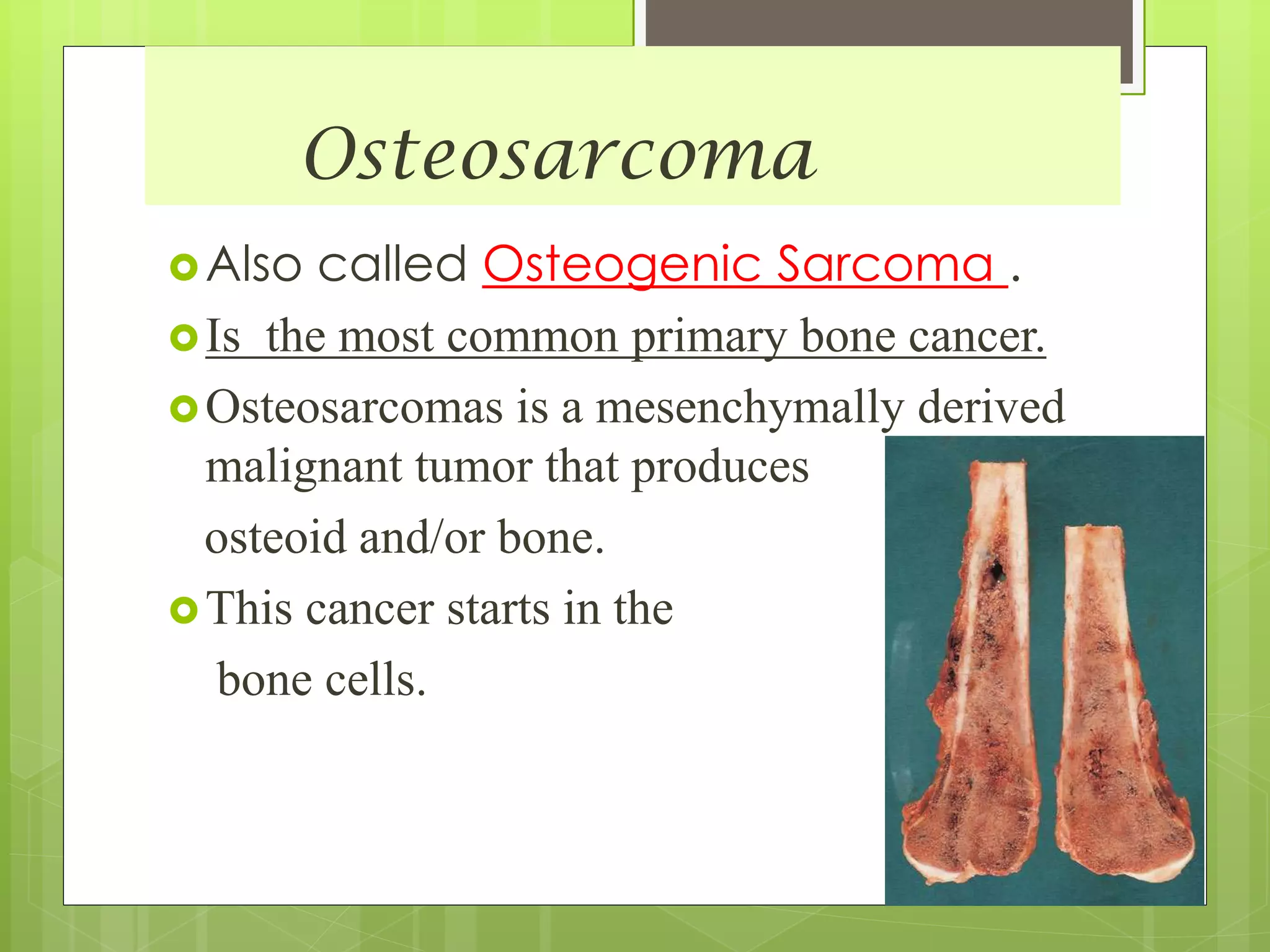
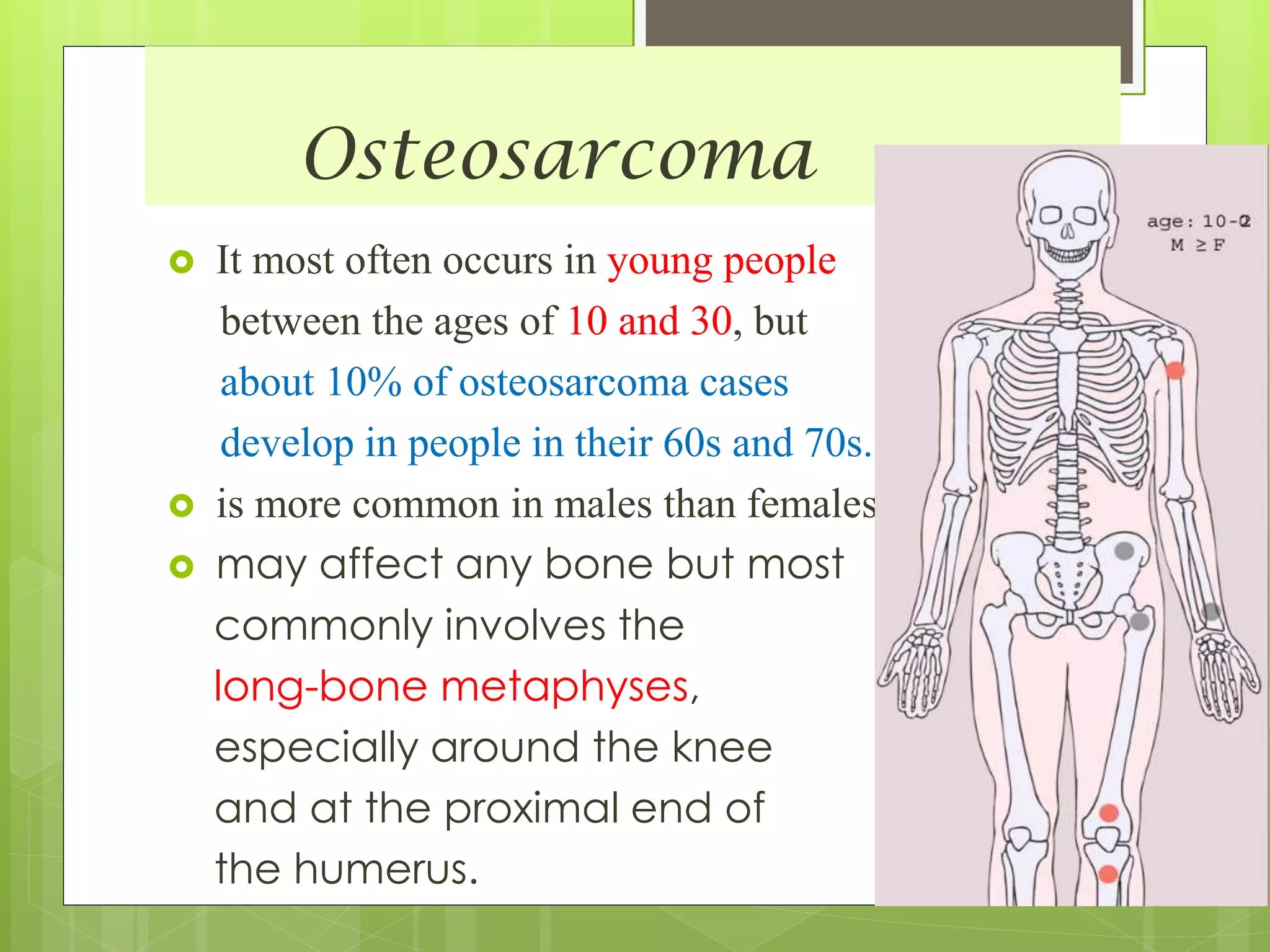

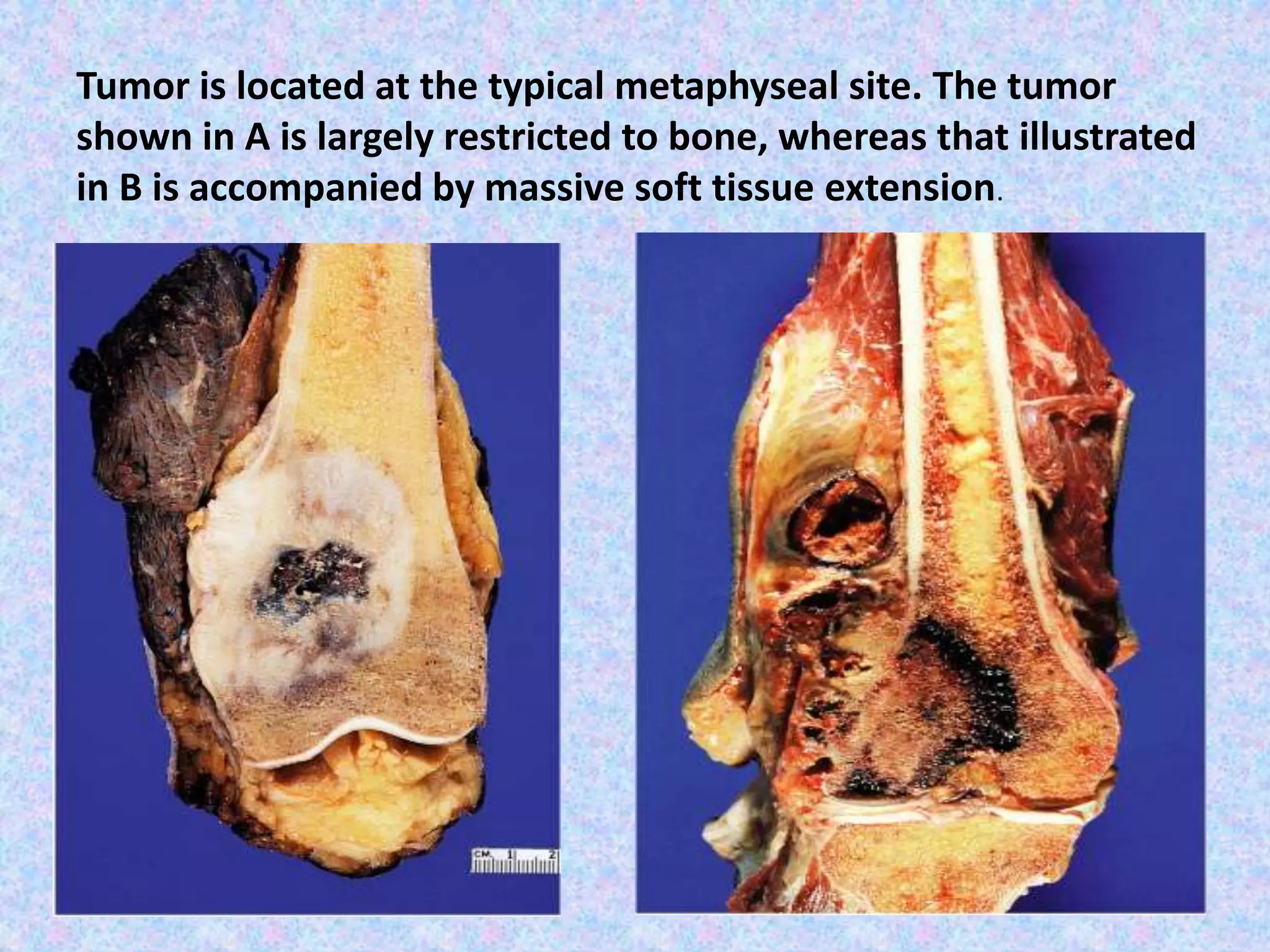




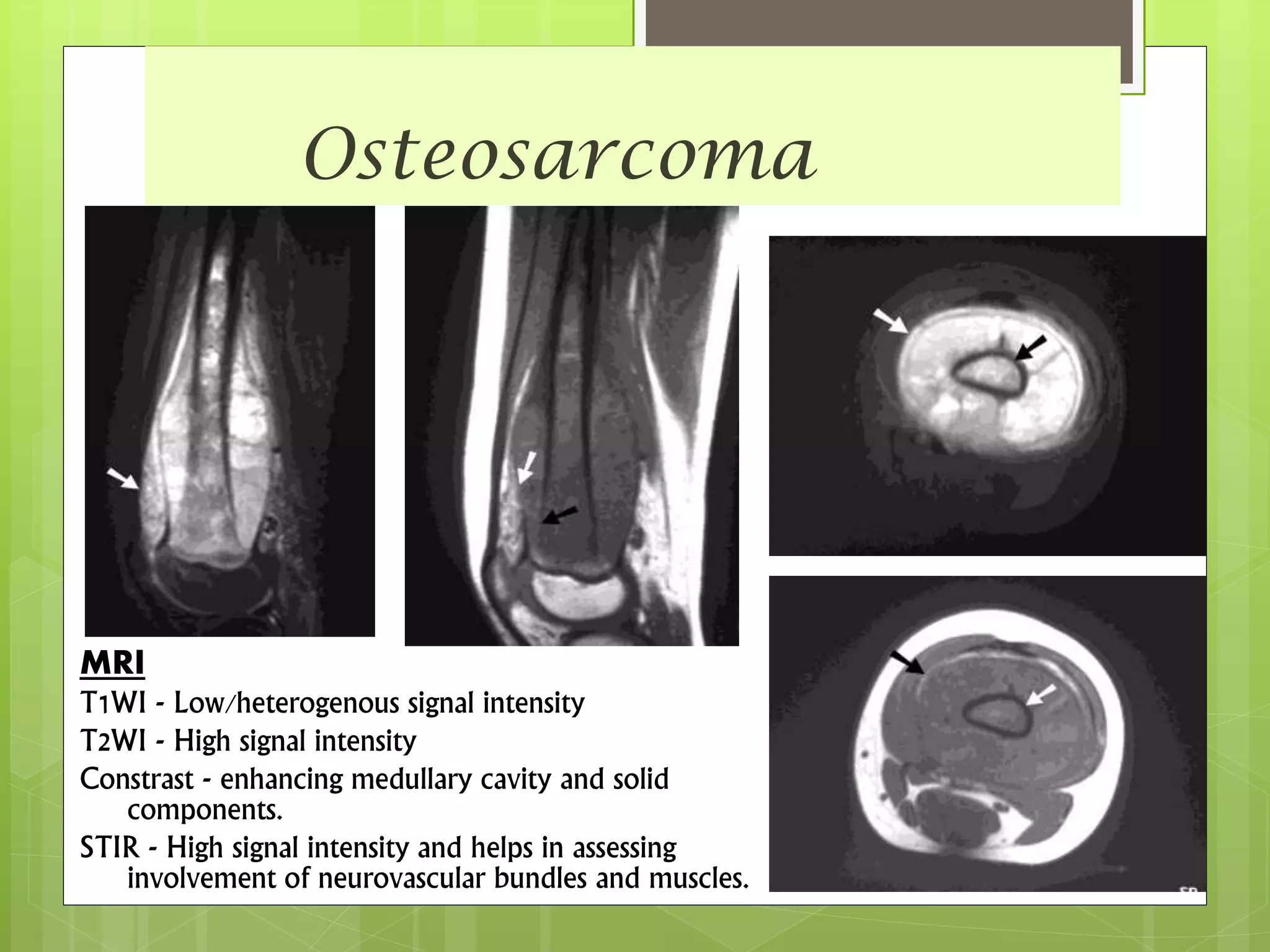
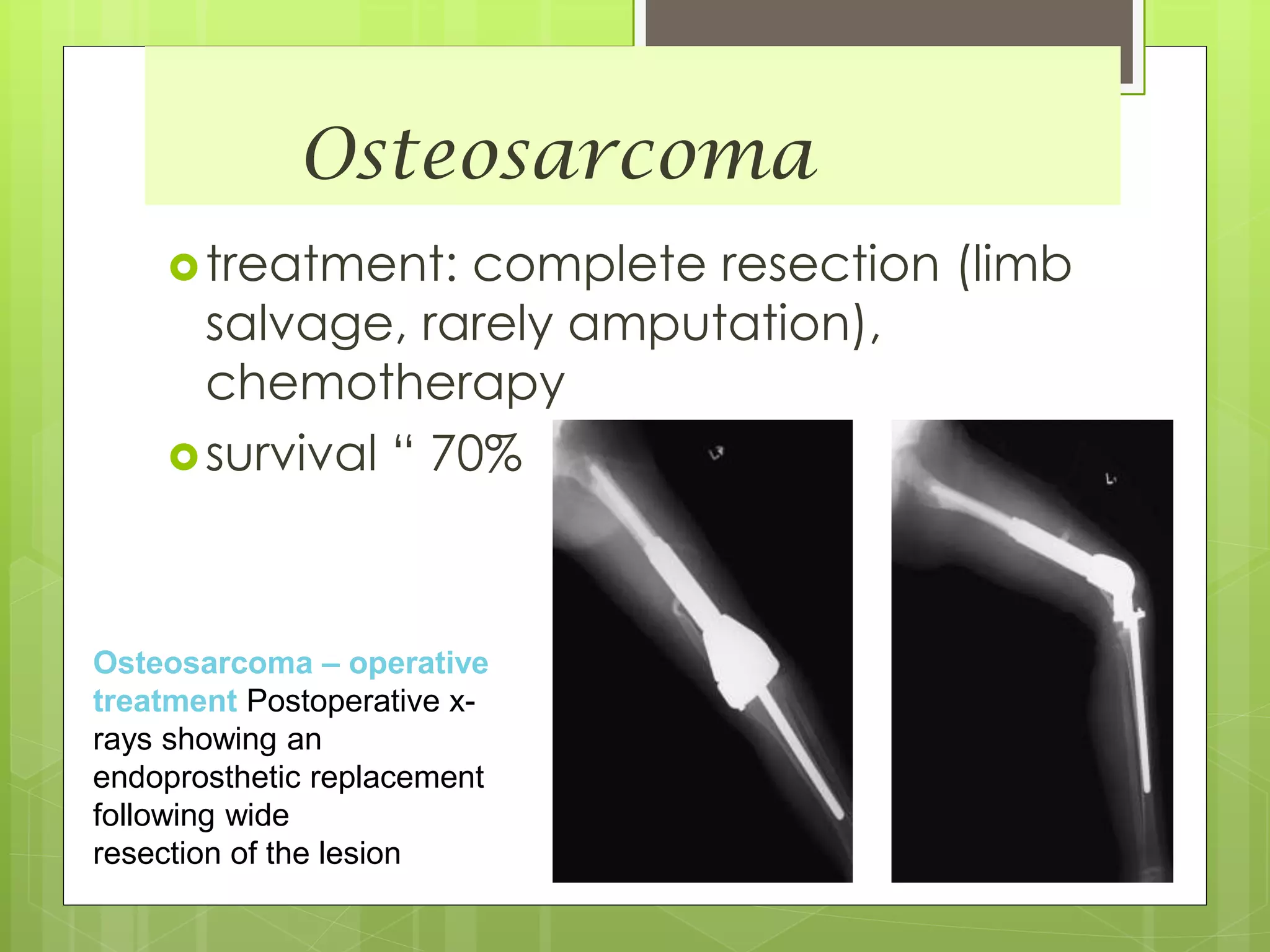








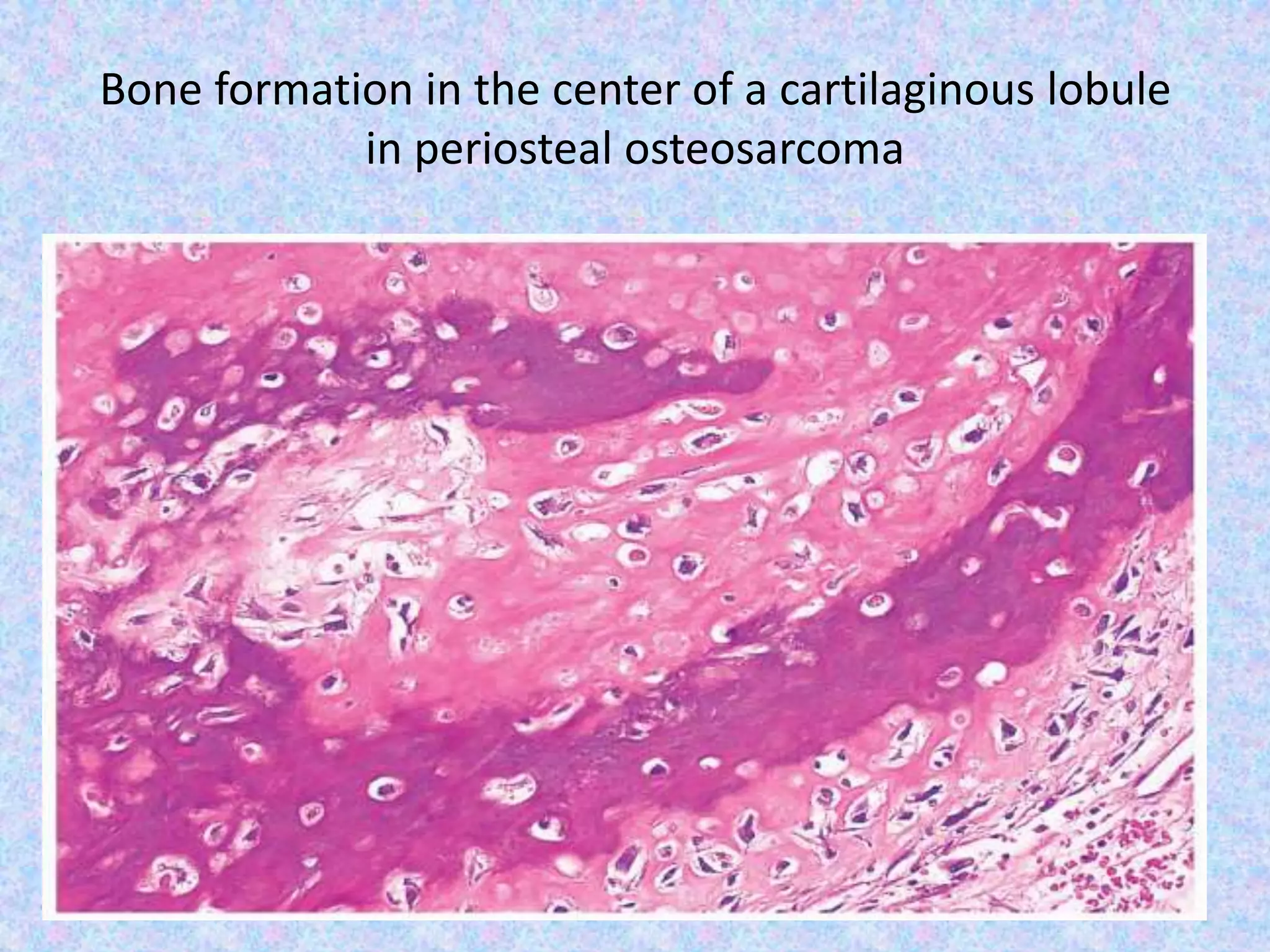


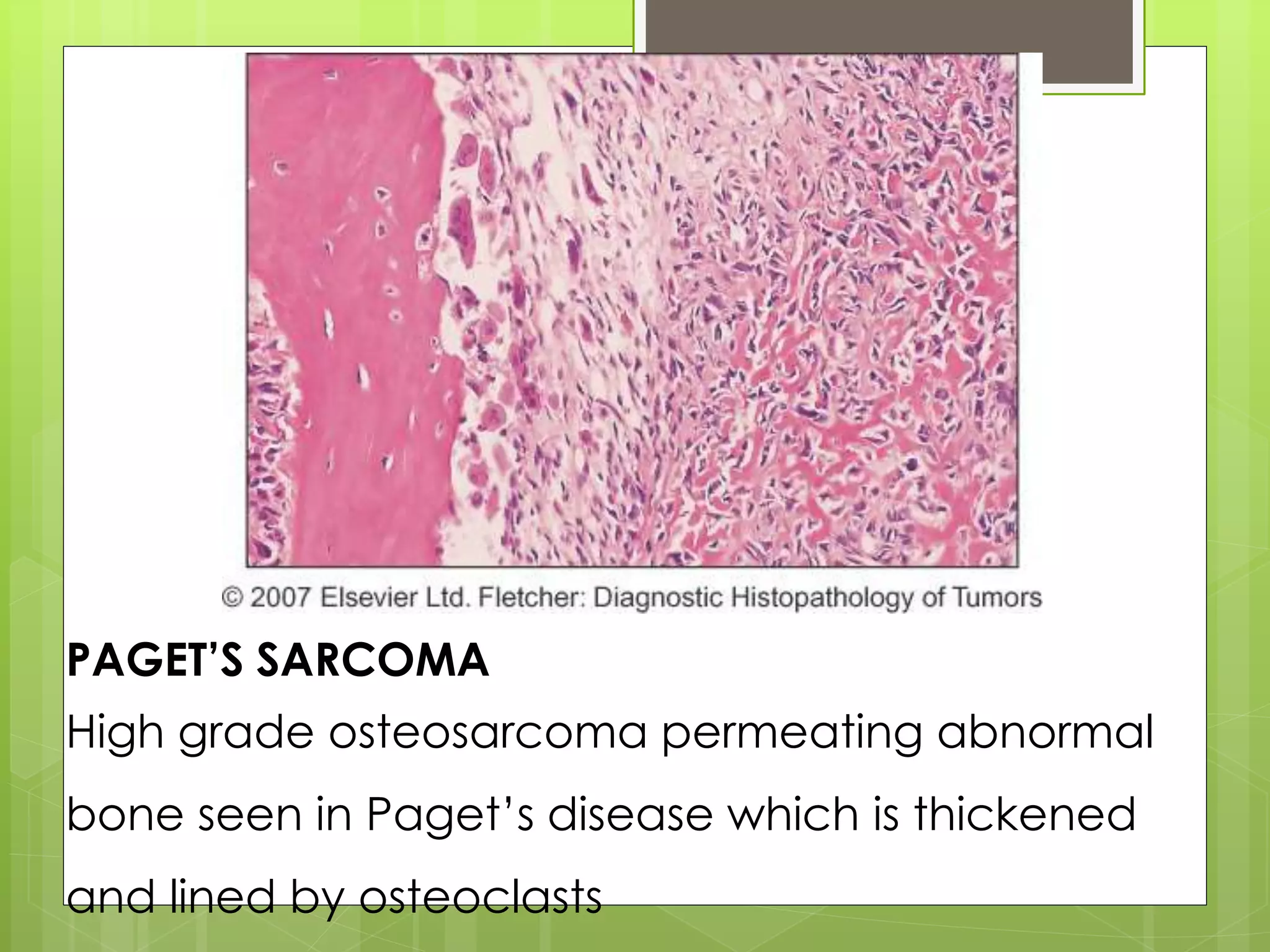





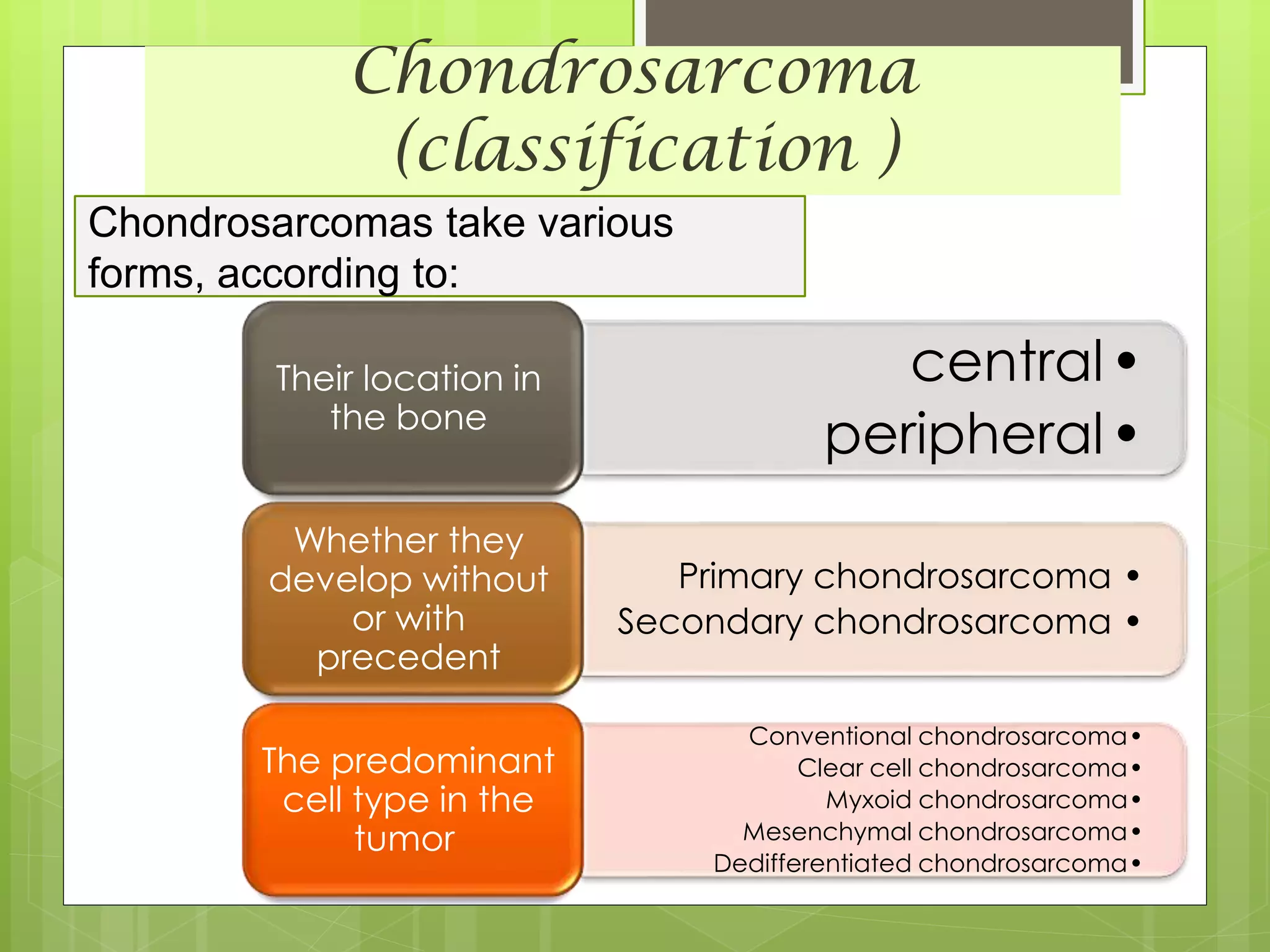



























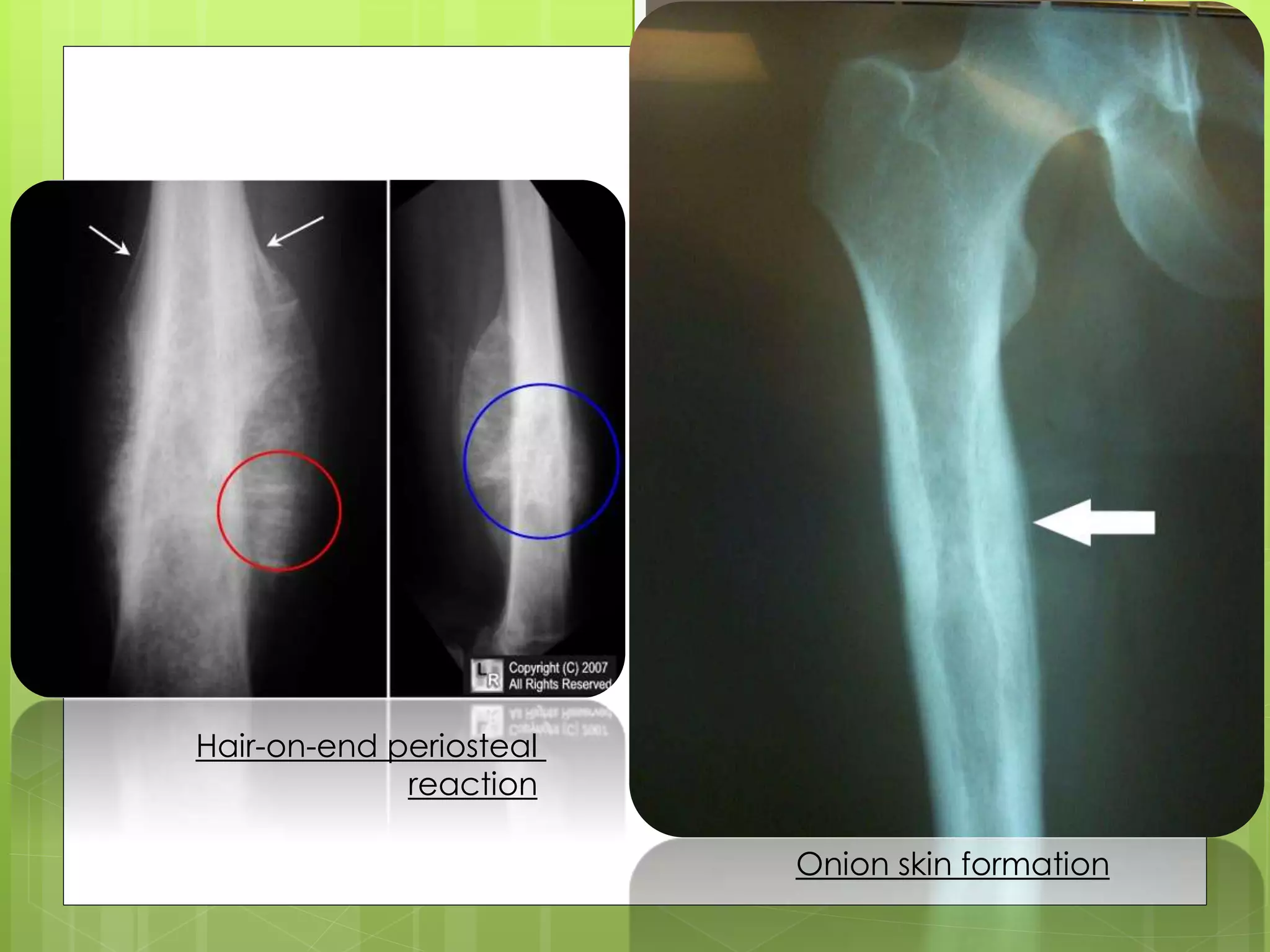













































The document provides a comprehensive overview of malignant bone tumors, including their classification, diagnosis, and treatment options. It discusses the different types of bone tumors, their epidemiology, risk factors, and the specific characteristics of tumors such as osteosarcoma and chondrosarcoma. Treatment approaches, including surgery and chemotherapy, are highlighted, along with the importance of accurate staging for prognosis and therapeutic strategy.











































































































