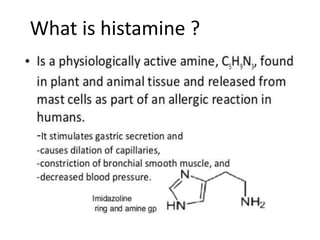
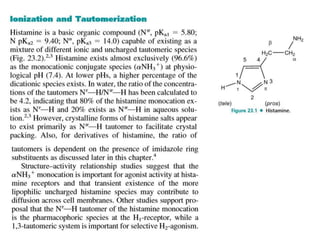
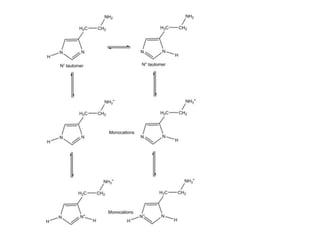
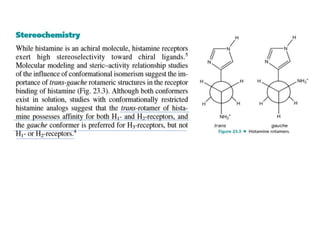
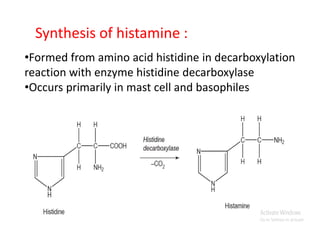
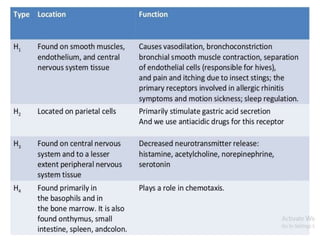
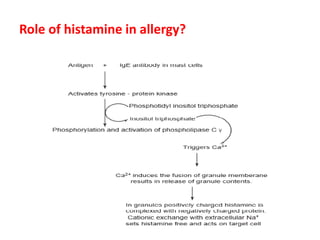
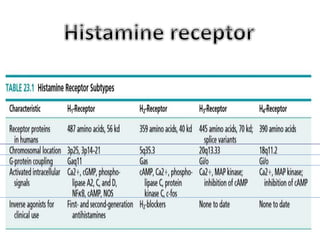
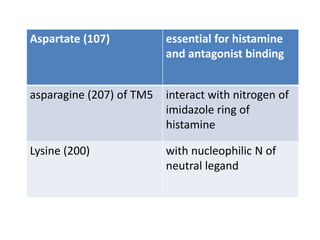
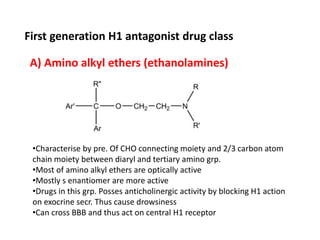
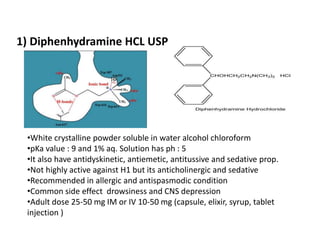
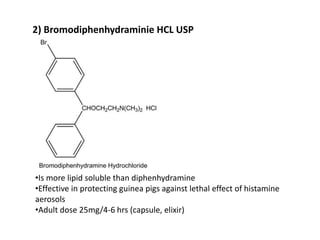
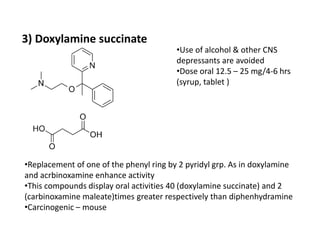
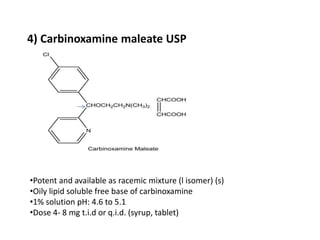
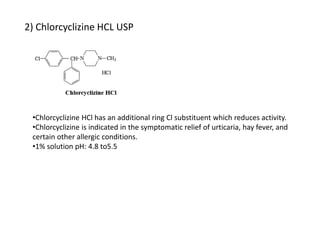
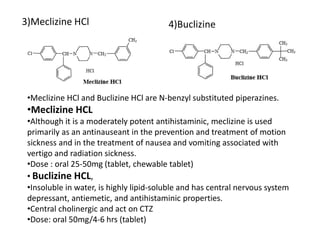
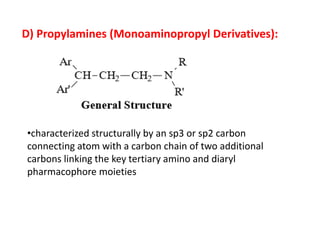
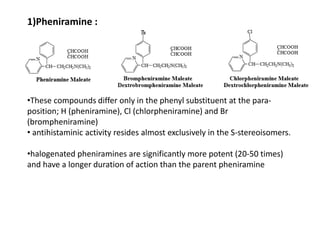
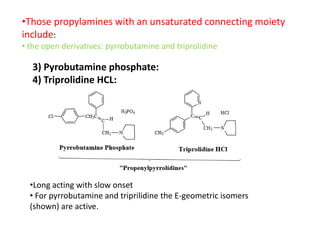
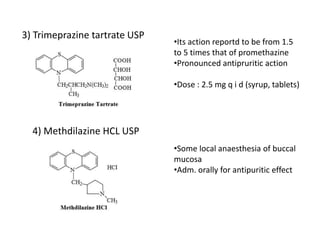
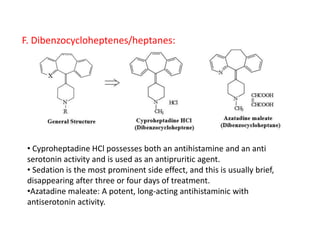
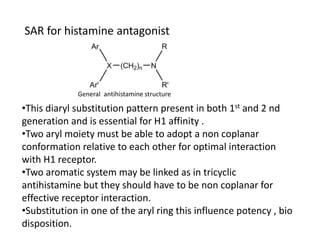
Antihistaminic agents are drugs that reduce or eliminate the effects of the chemical histamine by blocking its action on H1 receptors. There are several classes of antihistamines including amino alkyl ethers, ethylenediamines, piperazines, and propylamines. Within each class, specific drugs like diphenhydramine, cyclizine, chlorpheniramine differ in their chemical structure and properties like potency, duration of action, and side effects. All antihistamines work by competitively blocking the H1 receptor to inhibit the effects of histamine, providing relief for allergic conditions.