Download to read offline

![888 Am. J. Hum. Genet. 76:887–883, 2005
heterozygotes are 1, 3, 5,…, . When is even, pos-
n n
A A
sible numbers of heterozygotes are 0, 2, 4,…, . The
nA
expression for given in equation (1) leads
P(n FN,n )
AB A
to natural tests for HWE. For example, one could
define one-sided tests that focus on detection of a de-
ficit of heterozygotes, by calculating the statistic P p
low
, or detection of an excess of heter-
P(N ⭐ n FN,n )
AB AB A
ozygotes, by calculating the statistic P p P(N ⭓
high AB
. In each case, the statistic can be calculated
n FN,n )
AB A
by simply summing over equation (1), to include all pos-
sible values of that are lower (for ) or higher (for
N P
AB low
) than those observed in the actual data. A test for
Phigh
a deficit of heterozygotes in relation to Hardy-Weinberg
expectations is appropriate when deviations from HWE
due to inbreeding or population stratification are sus-
pected, since both of these increase the proportion of
homozygotes in the population. A test for an excess
of heterozygotes is appropriate when one suspects prob-
lems in genotyping due to the existence of highly ho-
mologous regions in the genome, since these low-copy
repeats often lead to an increase in the proportion of
apparent heterozygotes in the sample. In other settings,
it might be appropriate to use both tests. For example,
many technologies score genotypes by clustering signals,
and misspecified clusters can result in either vast excesses
or vast deficits of heterozygotes.
When neither an increase nor a decrease in the pro-
portion of heterozygotes is specifically expected, one
could perform two separate one-sided tests or, instead,
use a two-sided test statistic (Weir 1996). A natural
two-sided test statistic could be defined as P p
2a
. This two-sided statistic is appeal-
min (1.0, 2P , 2P )
high low
ing because it leads to rejection of HWE at significance
level 2a in instances in which the one-sided tests lead to
the rejection of HWE at significance level a. However,
because of the asymmetric nature of the distribution of
heterozygote counts in a sample, the statistic is quite
conservative in practice, and we do not recommend
its use. Instead, an appealing approach, analogous to
Fisher’s exact test for contingency tables (Fisher 1934),
is to calculate the probability of observing a sample con-
figuration that is even less likely than the one being eval-
uated, conditional on the observed allele counts. This
can be achieved using a statistic similar to the Monte
Carlo statistic proposed by Guo and Thompson (1992)
for multiallelic markers:
P p I P(N p n FN,n )
[
冘
HWE AB AB A
∗
nAB
∗
⭓ P(N p n FN,n ) ]
AB AB A
∗
#P(N p n FN,n ) .
AB AB A
In this definition, I[x] is an indicator function that is
equal to 1 when the comparison is true and equal to 0
otherwise. The sum should be performed over all het-
erozygote counts that are compatible with the ob-
∗
nAB
served number of minor alleles, .
nA
Most of the computational effort required for per-
forming exact tests of linkage disequilibrium is spent
evaluating the factorials in equation (1) for each possible
value of . By use of a naive approach, evaluating
nAB
equation (1) requires 5N–6N multiplications and one
division for each possible value of . We simplify cal-
nAB
culations by using the recurrence relationships previ-
ously recognized by Guo and Thompson (1992) in the
implementation of their Markov chain–Monte Carlo
sampler:
P(N p n ⫹ 2FN, n )
AB AB A
4n n
AA BB
p P(N p n FN, n ) , and
AB AB A
(n ⫹ 2)(n ⫹ 1)
AB AB
P(N p n ⫺ 2FN, n )
AB AB A
n (n ⫺ 1)
AB AB
p P(N p n FN, n ) . (2)
AB AB A
4(n ⫹ 1)(n ⫹ 1)
AA BB
In this way, evaluating the probability for each possible
number of heterozygotes takes only four multiplications
and one division, whatever the sample size N. To avoid
underflow, it is best to first calculate the probability of
observing the expected number of heterozygotes (in this
case, the most likely outcome) and then use the recur-
rence relationships to calculate probabilities for all other
outcomes. A further reduction of computational effort
is possible by noting that one need only calculate relative
probabilities for each outcome and then scale these to
ensure that their sum is 1.0. This means that the prob-
ability of observing the expected number of heterozy-
gotes can be replaced with an arbitrary constant when
using the recurrence relations in equation (2), provided
that the final result is scaled.
Table 1 illustrates the performance of the statistics for
a sample of 100 individuals in which 21 copies of the
minor allele are present. The observed number of het-
erozygotes will vary from 1 to 21 and must be odd. Note
that only a small number of distinct sample configura-
tions are possible, and each of these is associated with
a specific probability for the exact tests. If the desired
significance level a does not correspond exactly to one
of these discrete outcomes, then the exact test statistics
will be conservative (Hernandez and Weir 1989). For
example, at the significance level , the PHWE and
a p 0.05
Plow statistics both reject the hypothesis of HWE if ⭐13
heterozygotes are observed in this setting. Since the prob-
ability of observing ⭐13 heterozygotes is 0.010, the tests
are conservative. In contrast, the asymptotic x2
test sta-
tistic results in rejection of HWE when ⭐15 heterozy-](https://image.slidesharecdn.com/anoteonexacttestsofhardy-weinbergequilibrium-230804184546-24e1d73c/75/A-Note-On-Exact-Tests-Of-Hardy-Weinberg-Equilibrium-2-2048.jpg)
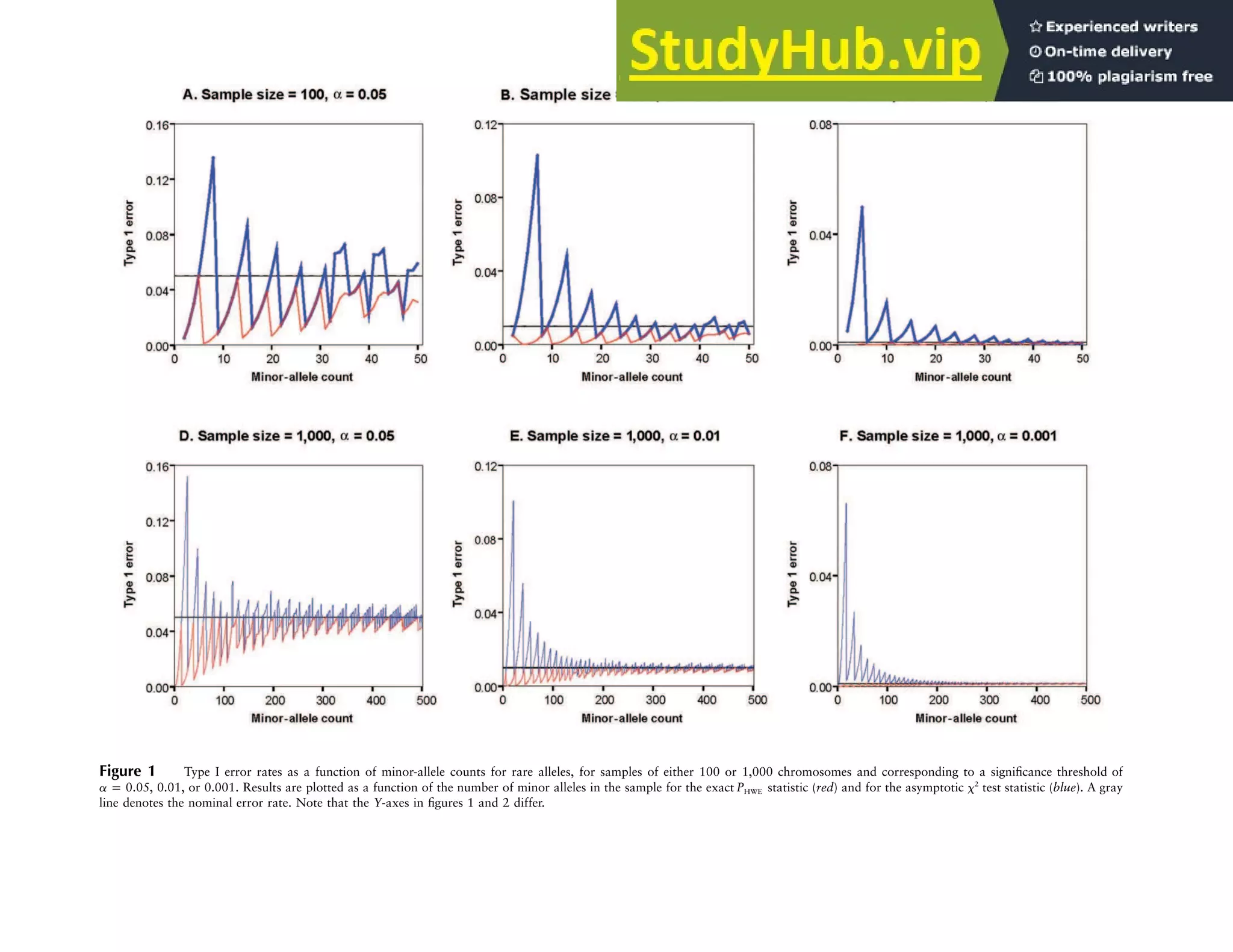

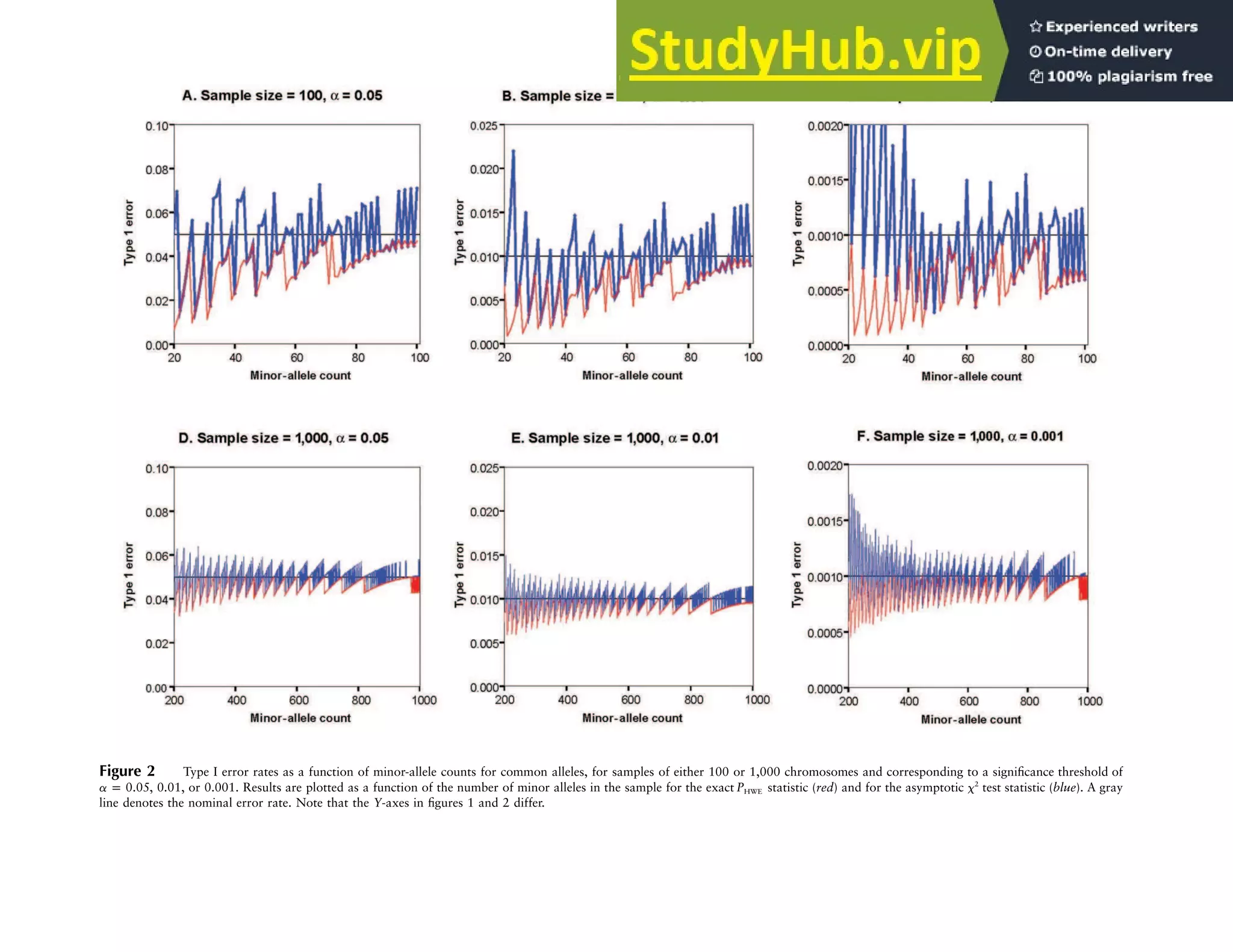



This document describes exact tests for assessing Hardy-Weinberg equilibrium that control type 1 error rates better than chi-squared tests, especially for large sample sizes. It presents efficient computational methods for implementing these exact tests, which sum probabilities of all possible genotype configurations conditional on observed allele counts. These exact tests have been programmed into freely available software for quality control and association testing in large genetic studies.



































































