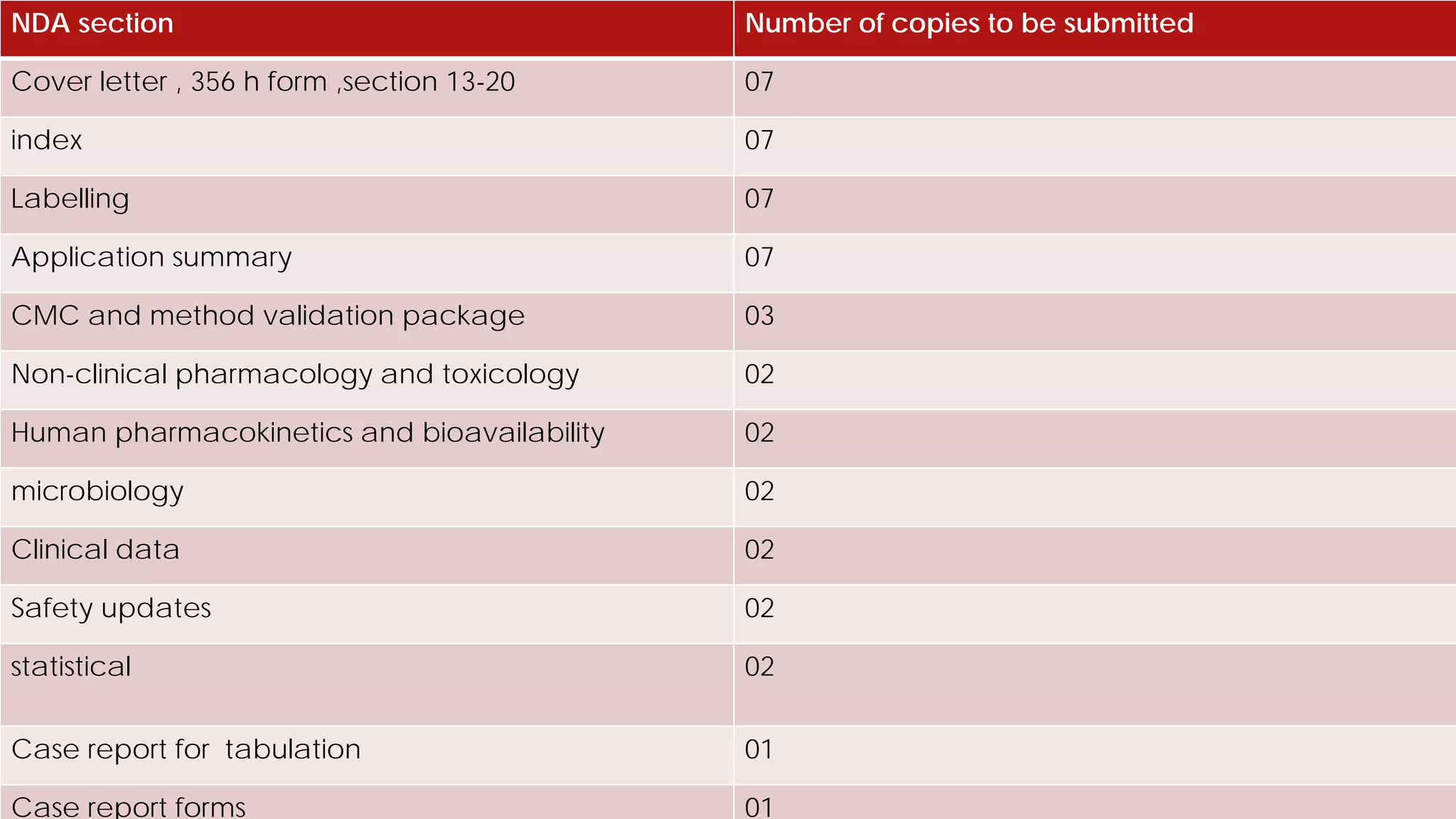
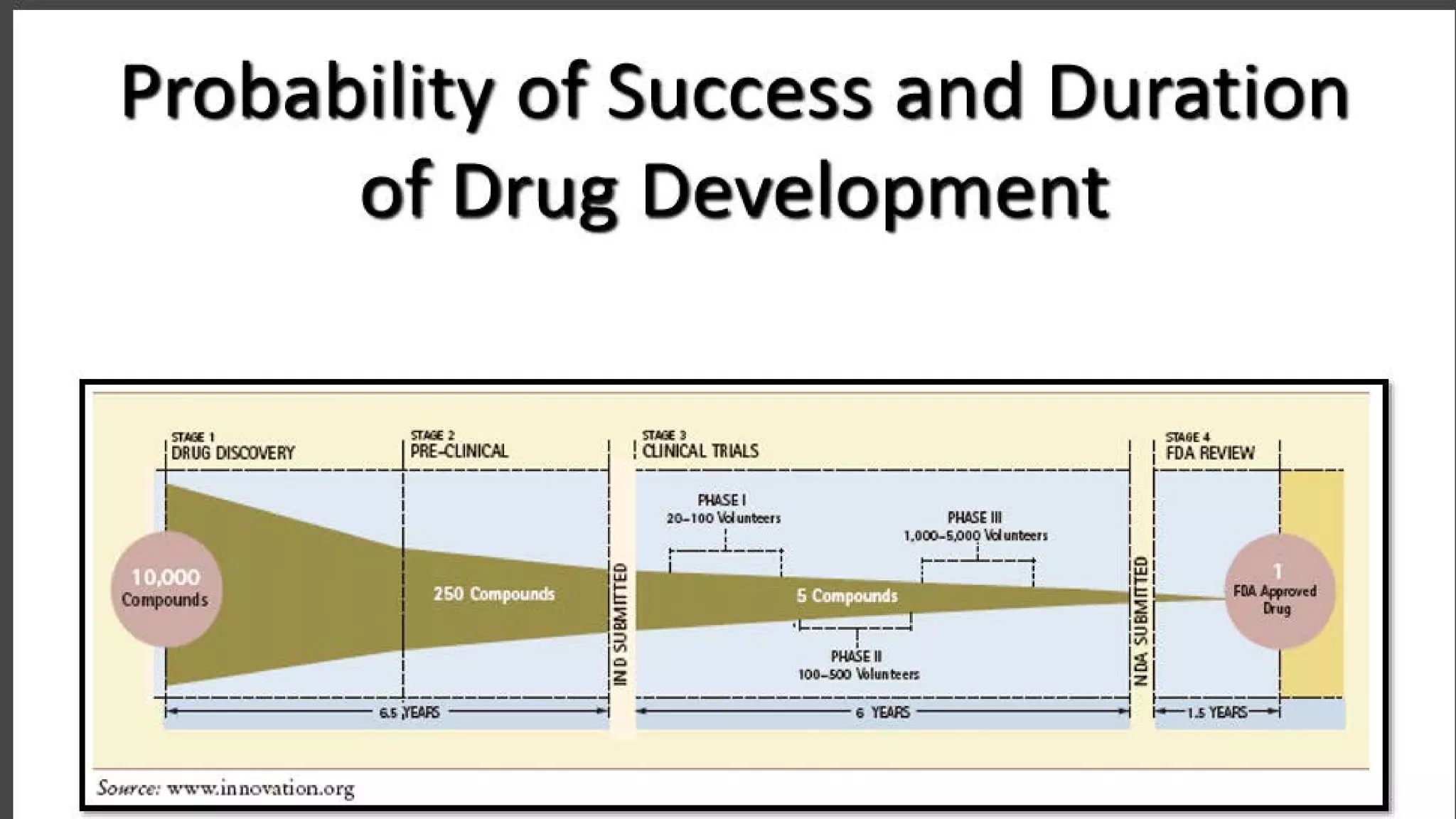
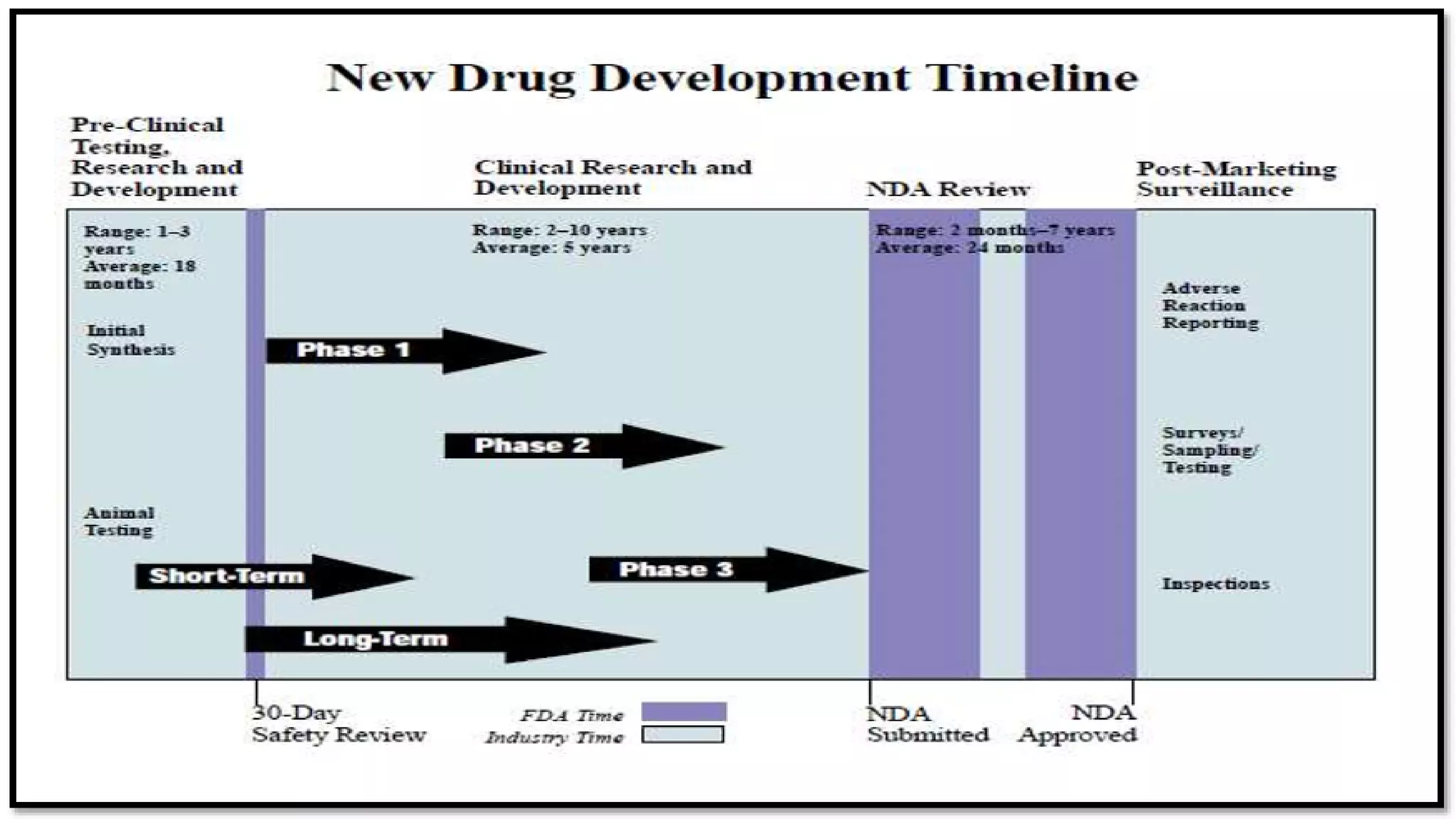
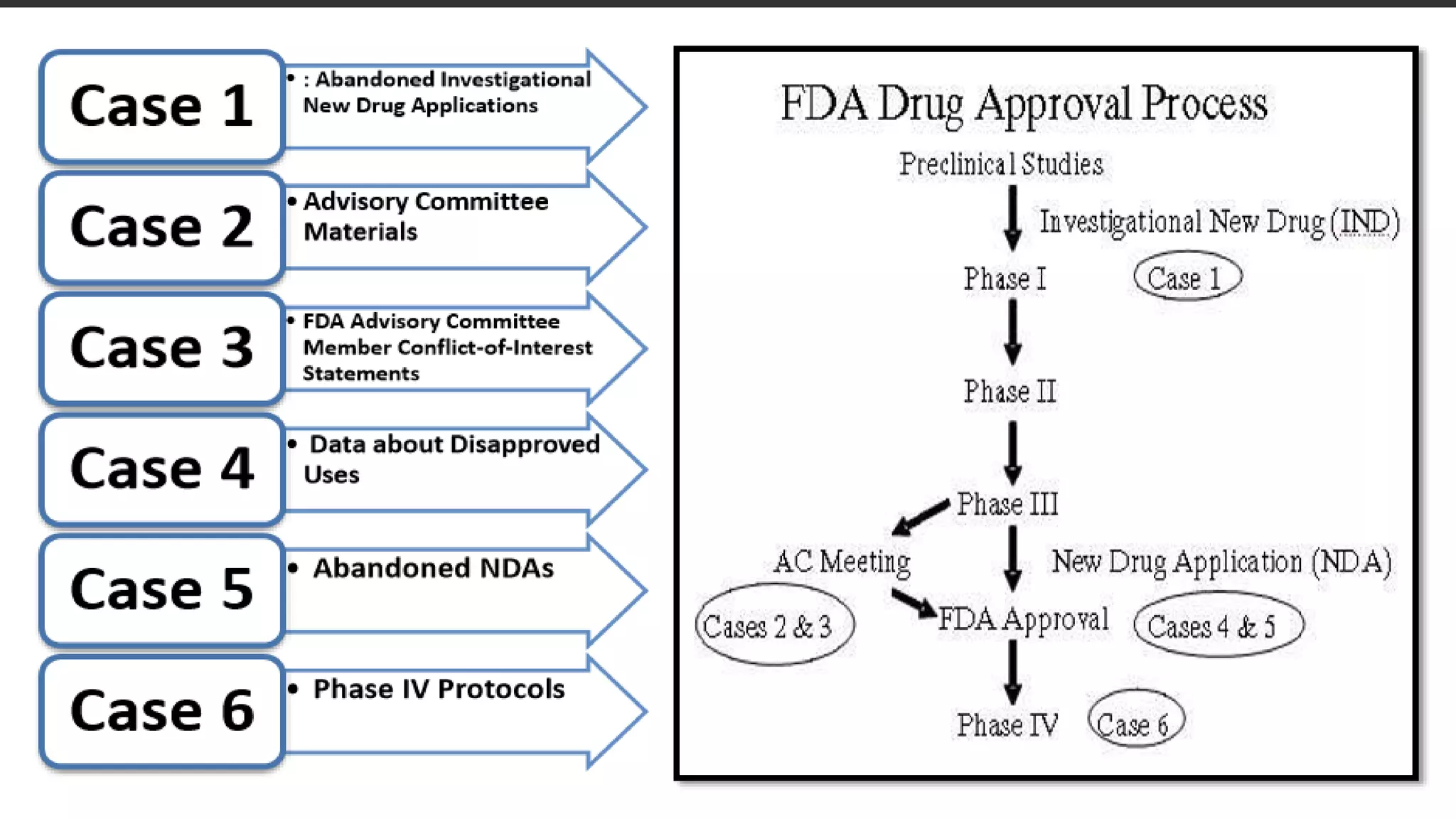
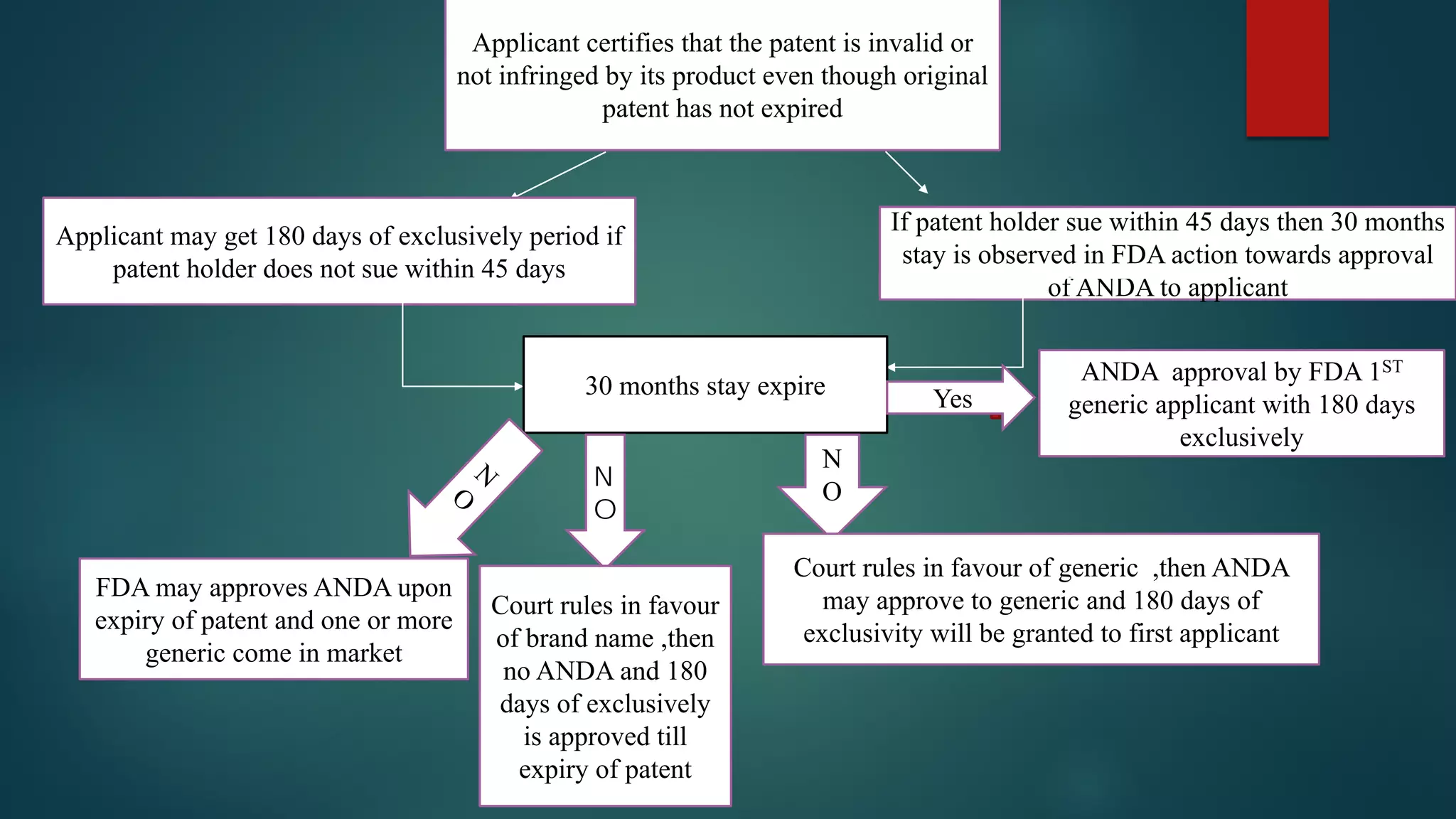
This document discusses the regulatory approval processes for new drug applications (NDAs) and abbreviated new drug applications (ANDAs) in the United States. It explains that an NDA contains extensive clinical and non-clinical data submitted by an innovator company to obtain approval for a new drug. An ANDA contains similar data to demonstrate equivalence to an already approved drug for generic versions after patents have expired. The document provides details on the requirements, review processes, and certification pathways for both NDAs and ANDAs.
















































![Abbreviated New Drug Application [ANDA]](https://cdn.slidesharecdn.com/ss_thumbnails/abbreviatednewdrugapplicationanda-160619062810-thumbnail.jpg?width=640&height=640&fit=bounds)







![New Drug Application [NDA]](https://cdn.slidesharecdn.com/ss_thumbnails/newdrugapplicationnda-160619063242-thumbnail.jpg?width=640&height=640&fit=bounds)
























