Download as PDF, PPTX




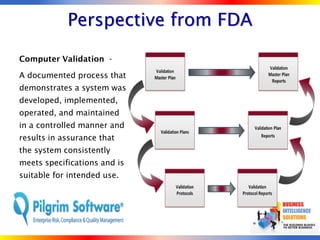













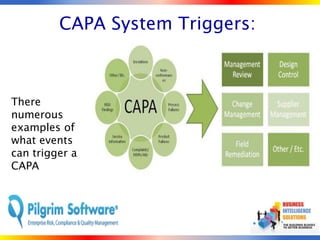


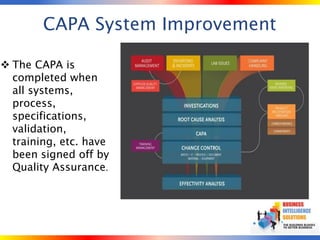




The document outlines strategies for computer system validation to achieve Part 11 compliance, focusing on guidelines and requirements from the FDA. It details processes such as Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ), as well as the importance of documentation and corrective action processes (CAPA). Additionally, it emphasizes the need for thorough audits and remediation activities to ensure effective validation management.






















































