Download as PDF, PPTX




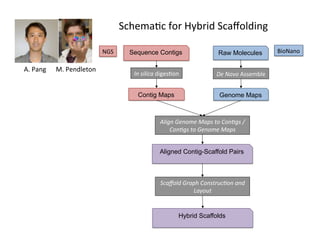
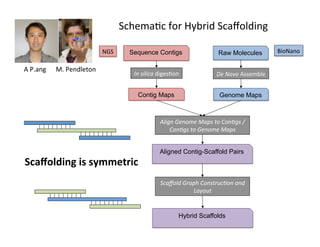
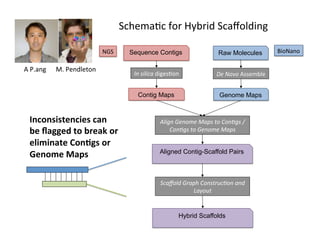

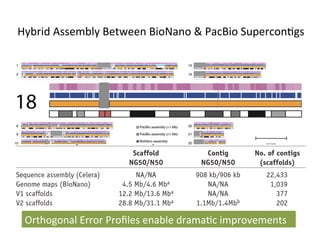
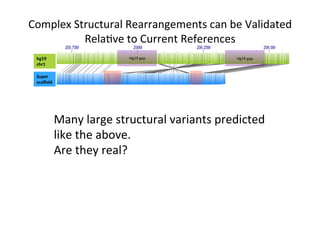
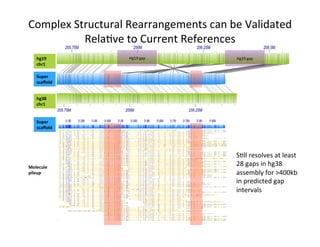
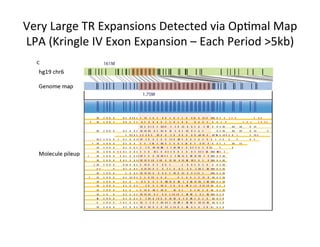
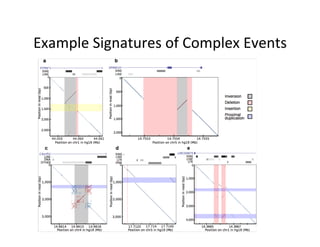
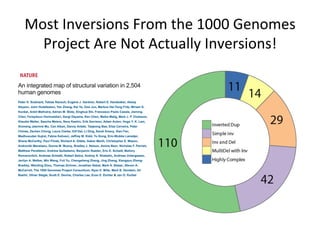
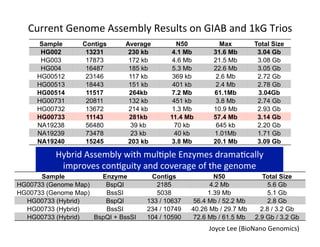
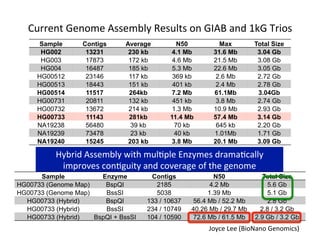
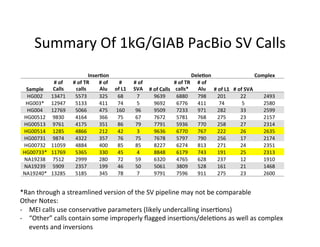


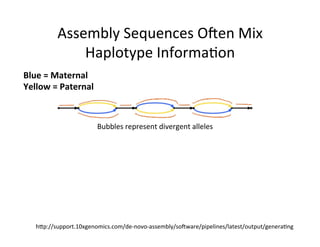
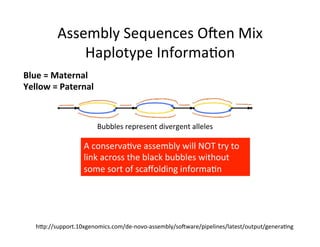
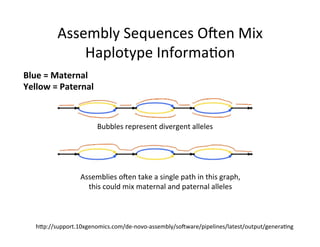
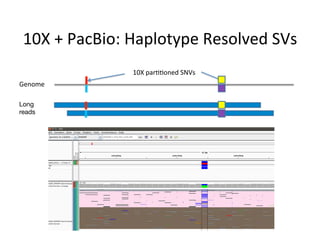

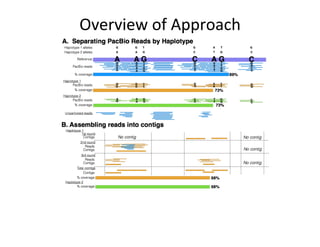

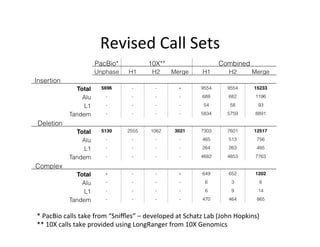

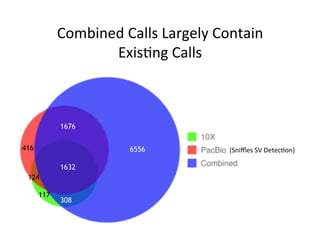
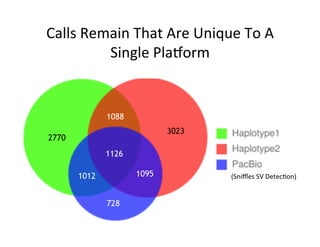
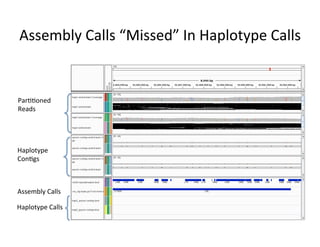
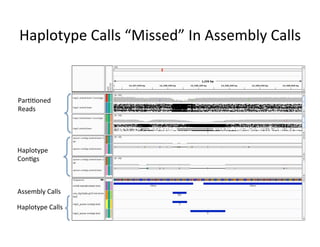
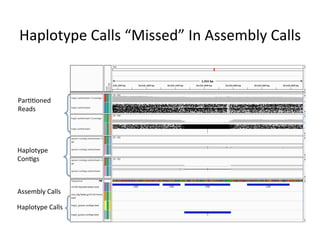



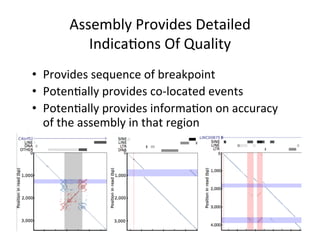
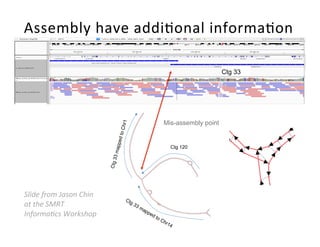



This document describes a method for haplotype-resolved structural variant assembly using long reads. PacBio and BioNano data are hybrid assembled to generate highly contiguous and complete haplotype-specific assemblies. The hybrid approach resolves many gaps in current references assemblies and detects complex structural variants and rearrangements. Analysis of trios from the 1000 Genomes Project and GIAB project using this pipeline detects numerous insertions, deletions, inversions and other structural variants.




















































































![ANIMAL_CELL_,_TISSUE_AND_ORGAN_CULTURE[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/animalcelltissueandorganculture1-260204172026-4462b440-thumbnail.jpg?width=640&height=640&fit=bounds)












