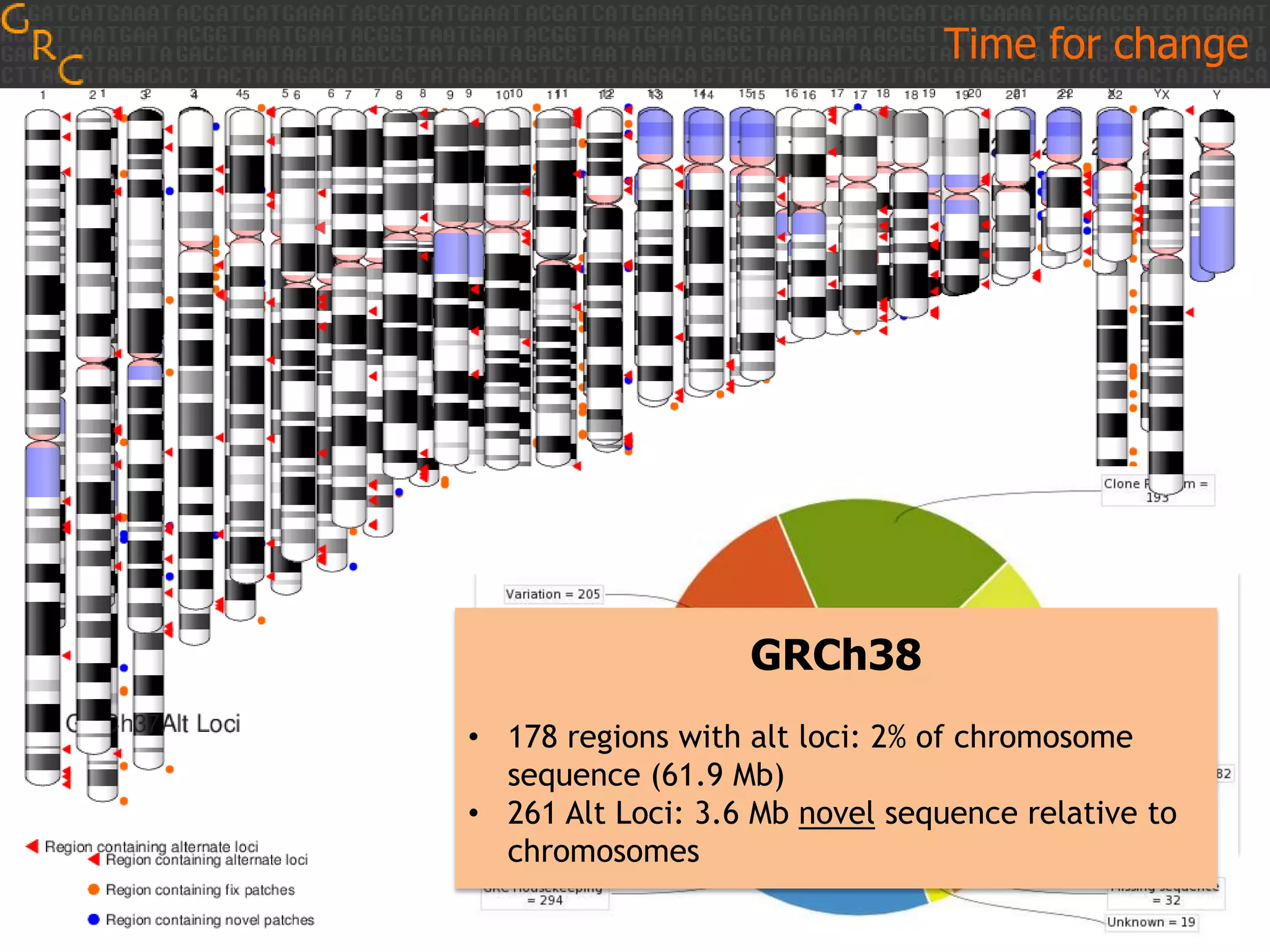
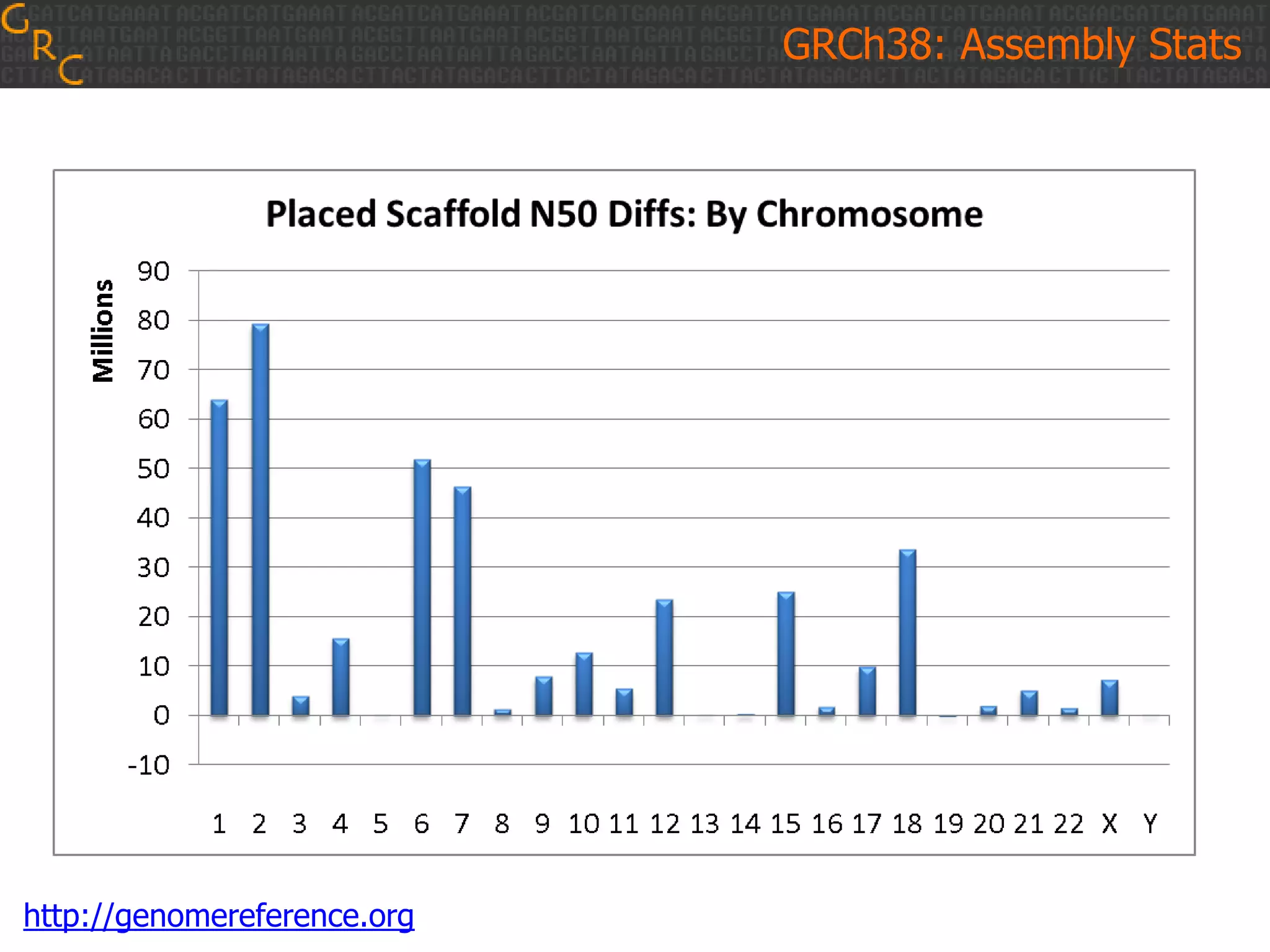
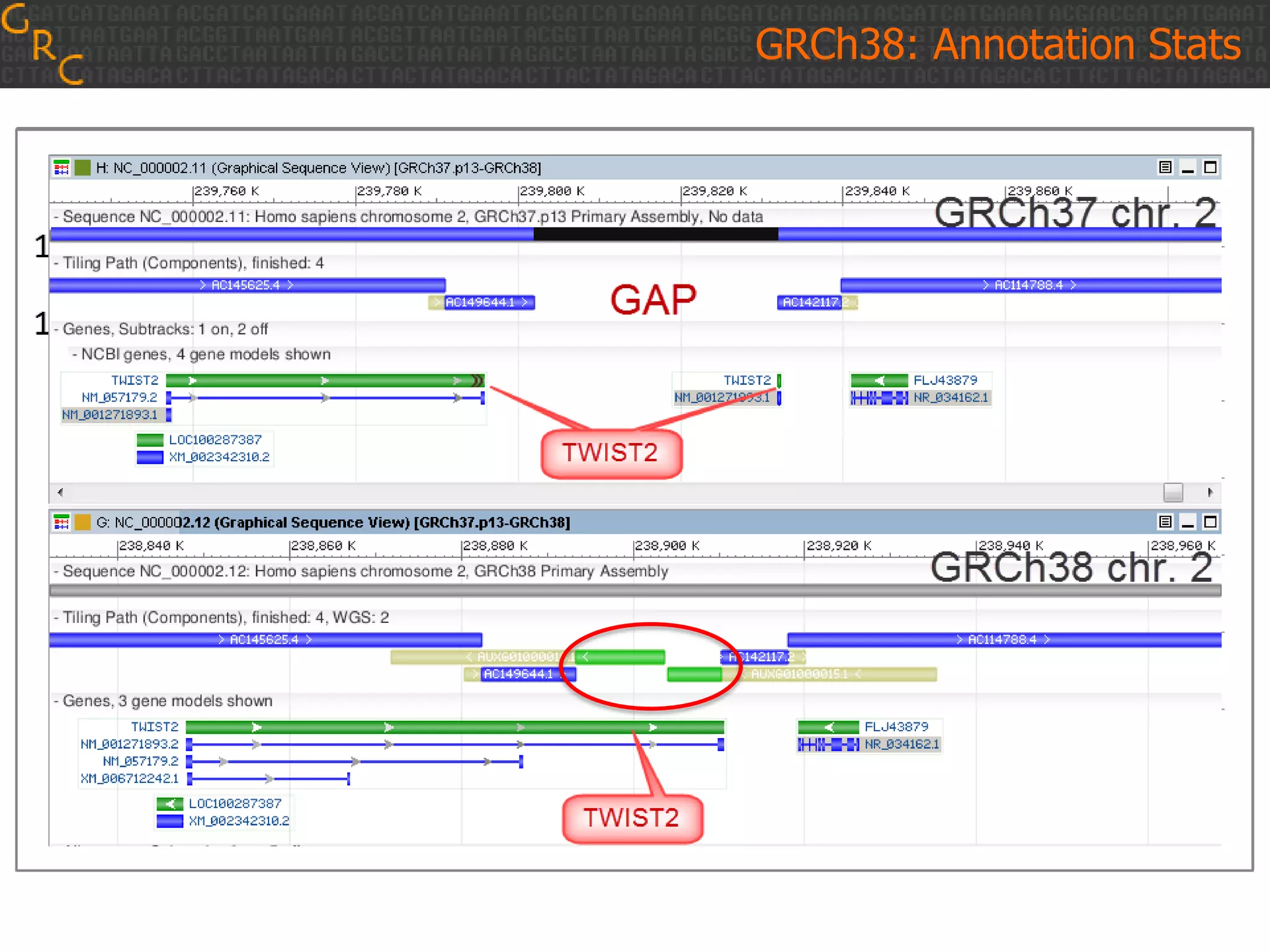
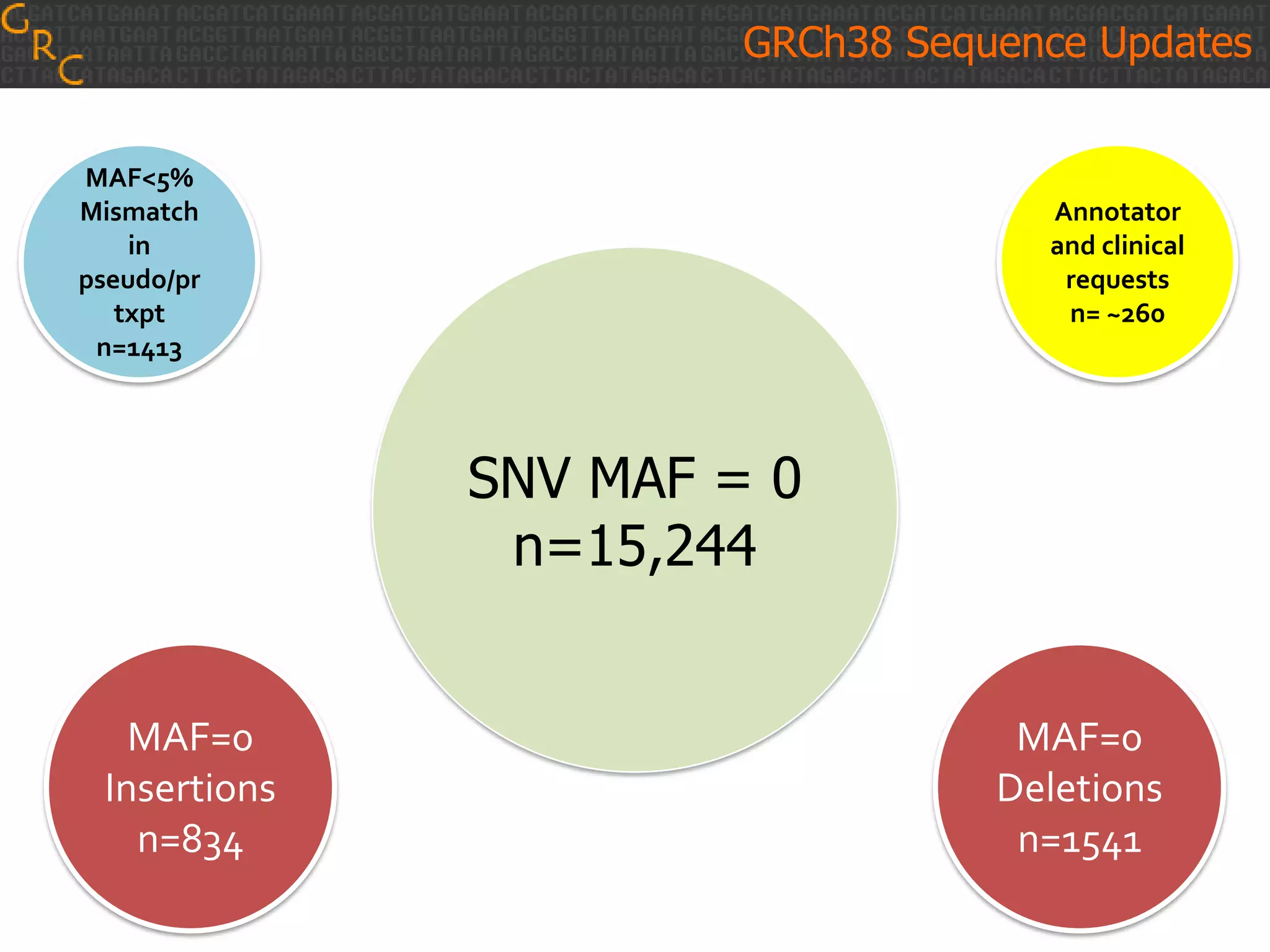
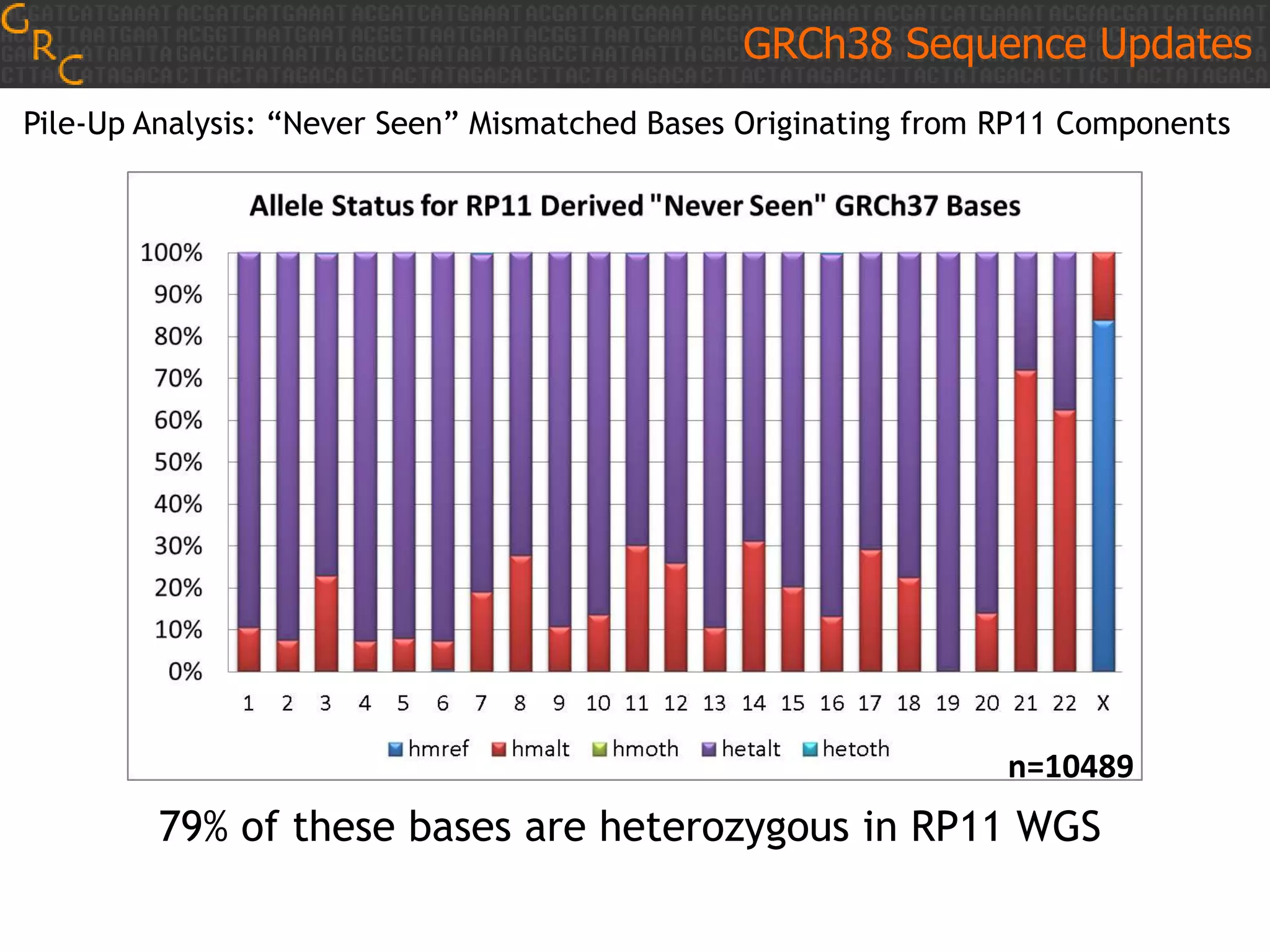
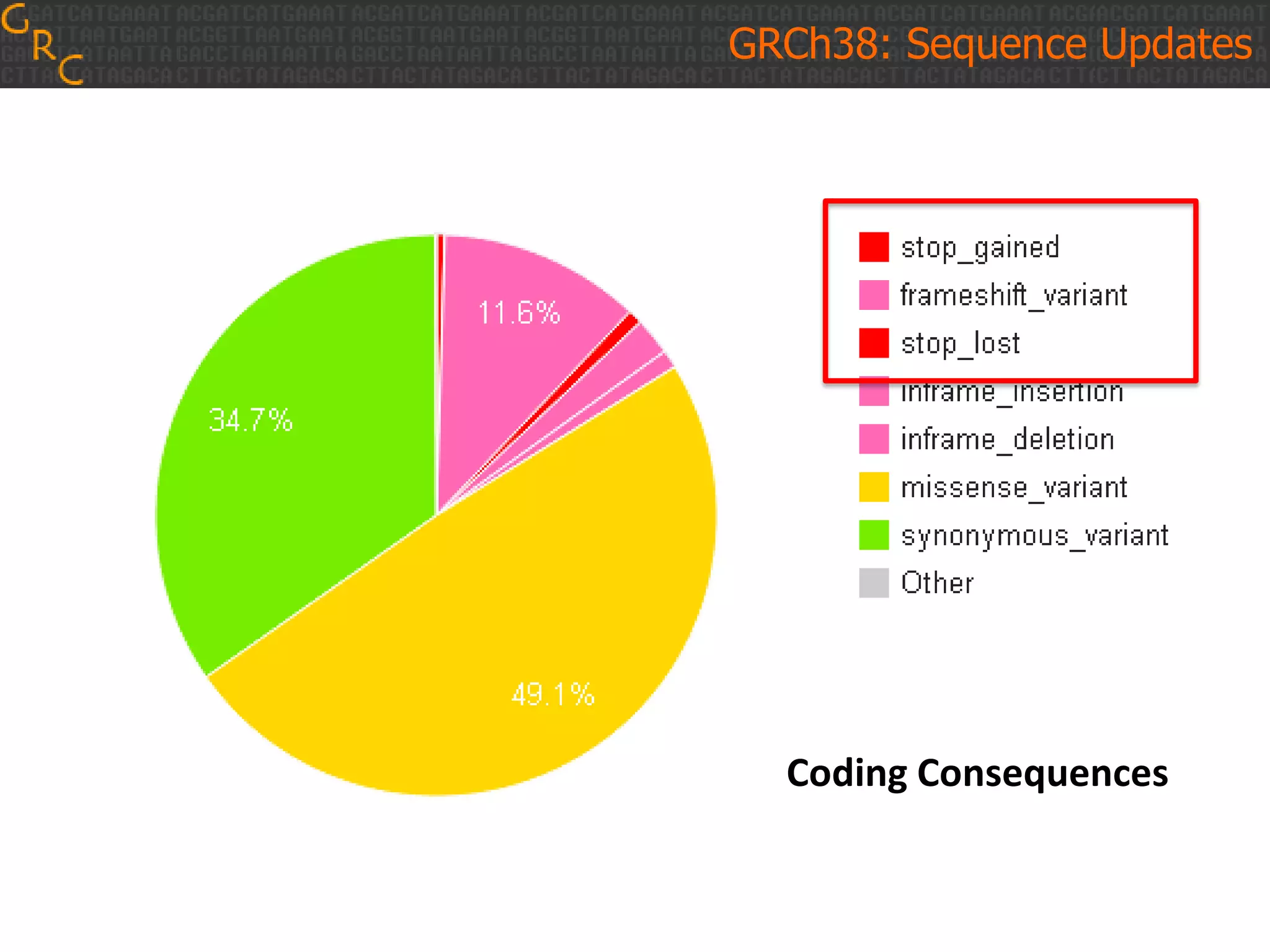
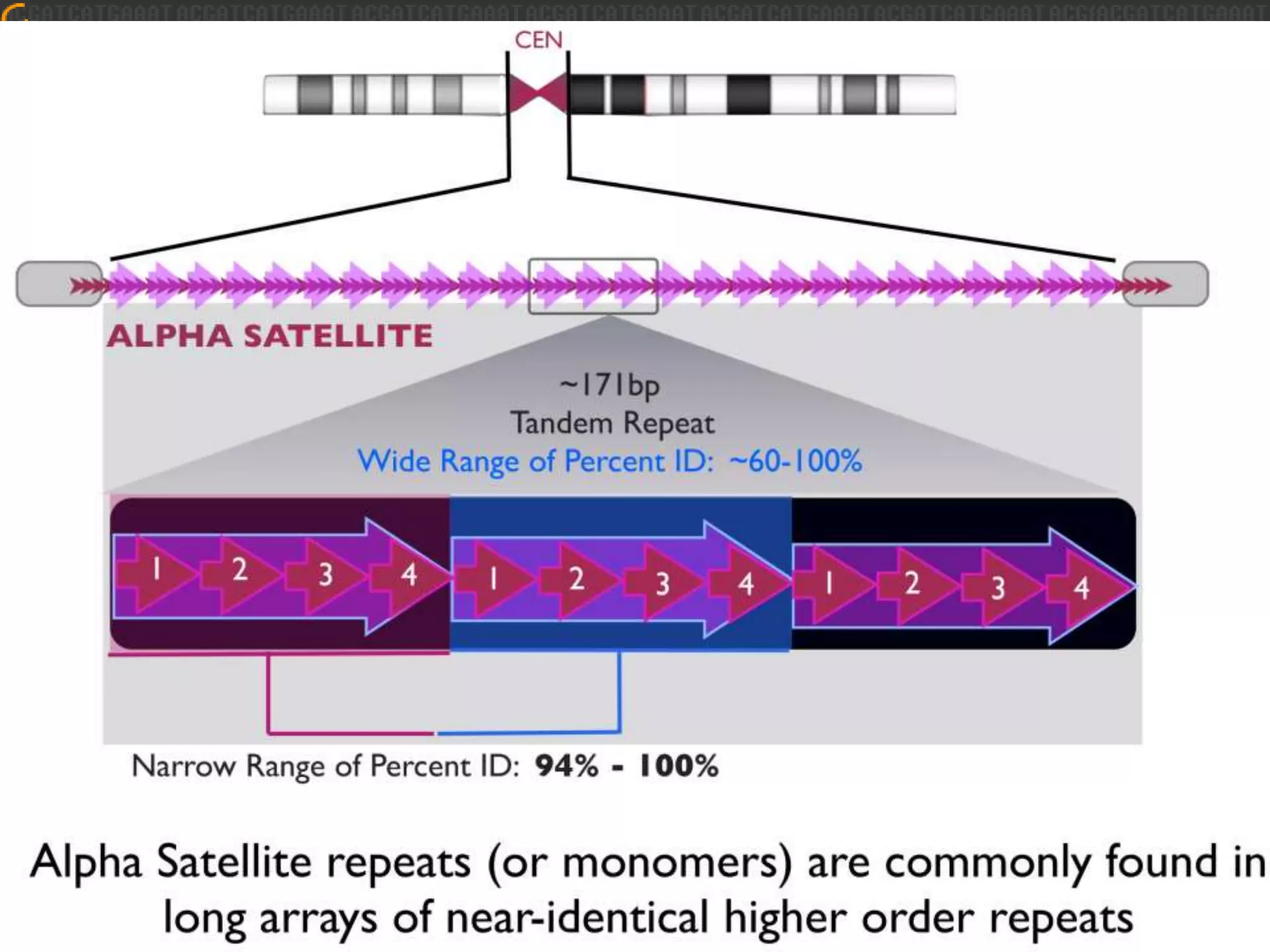
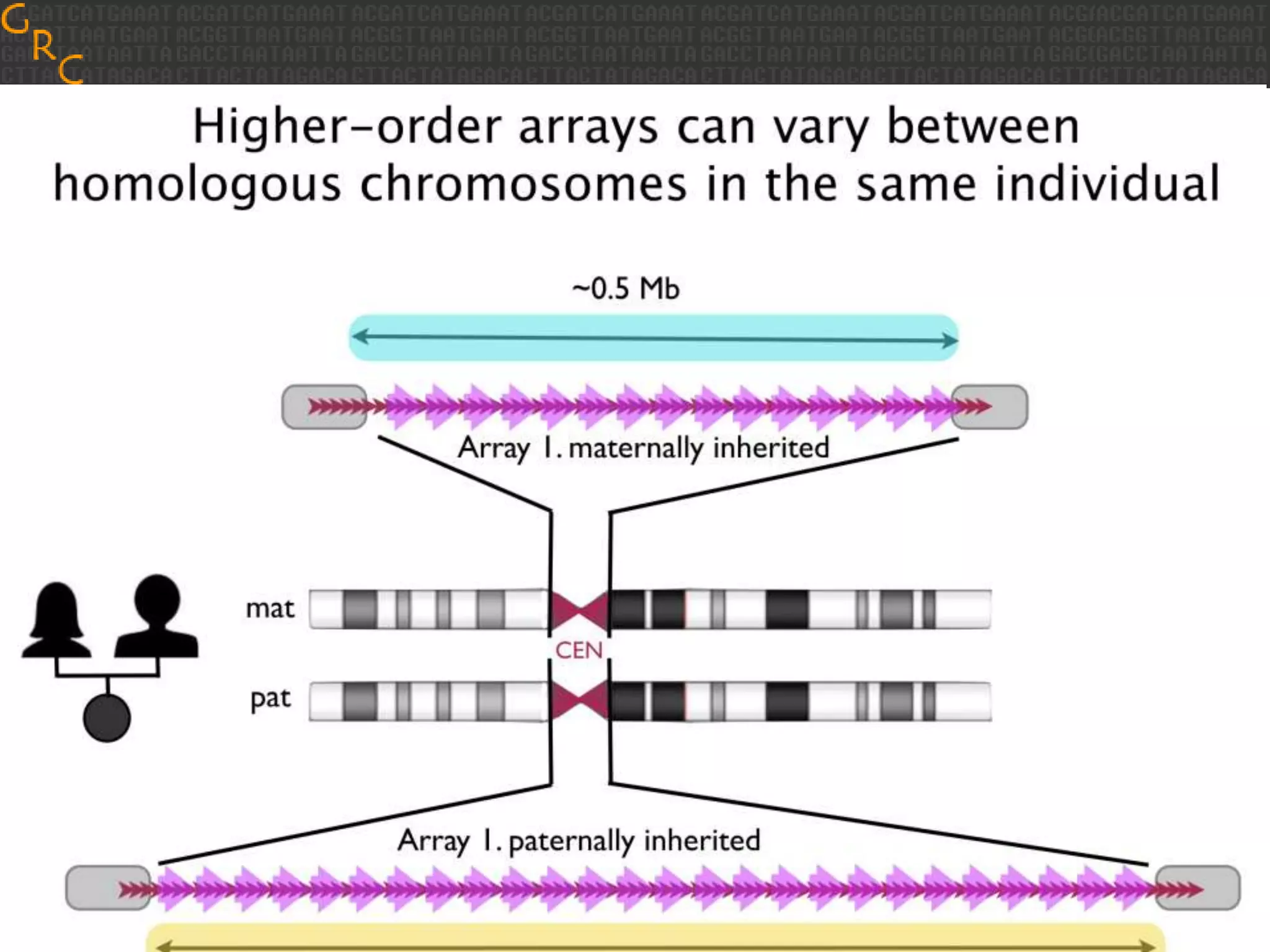
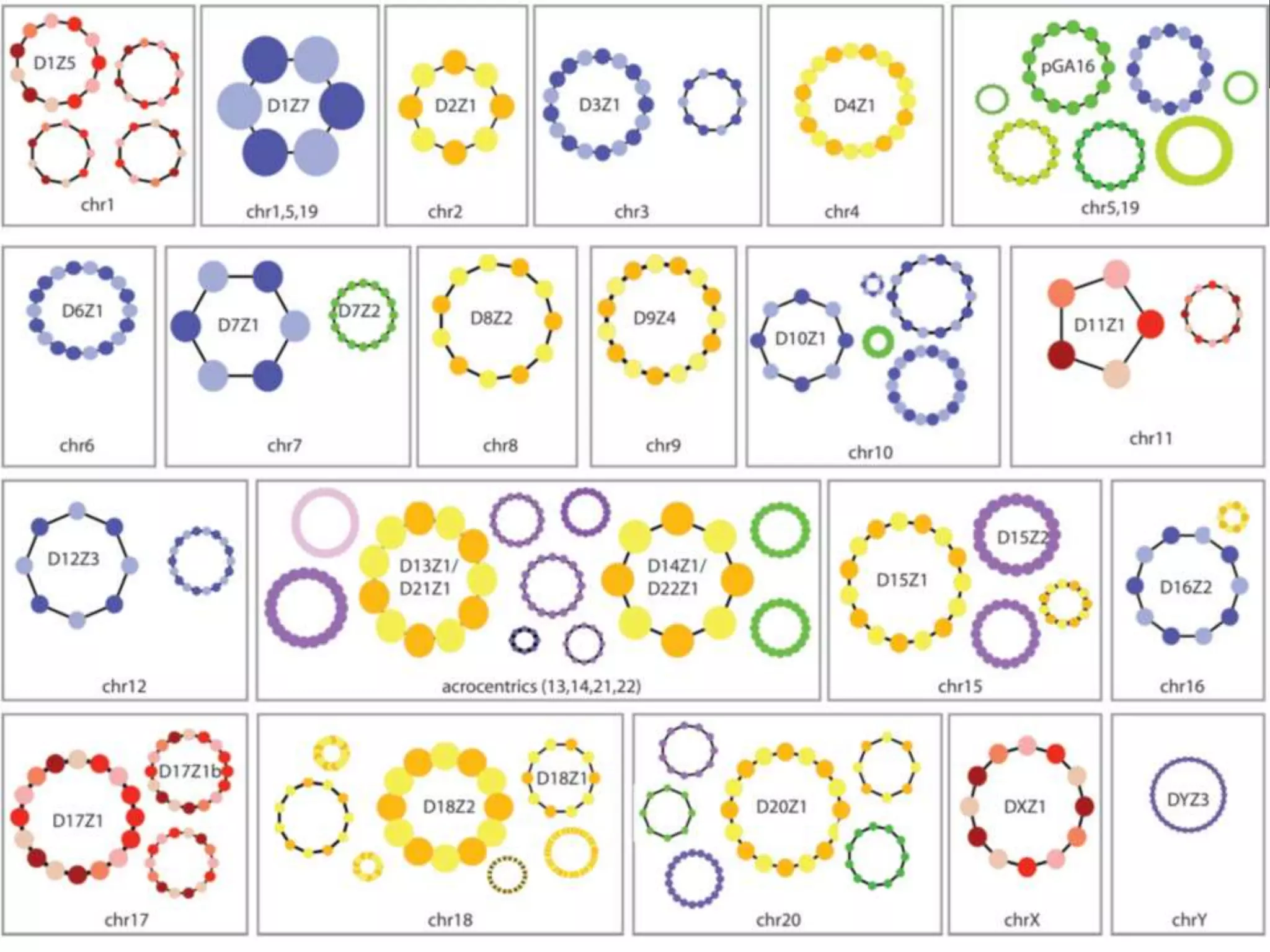
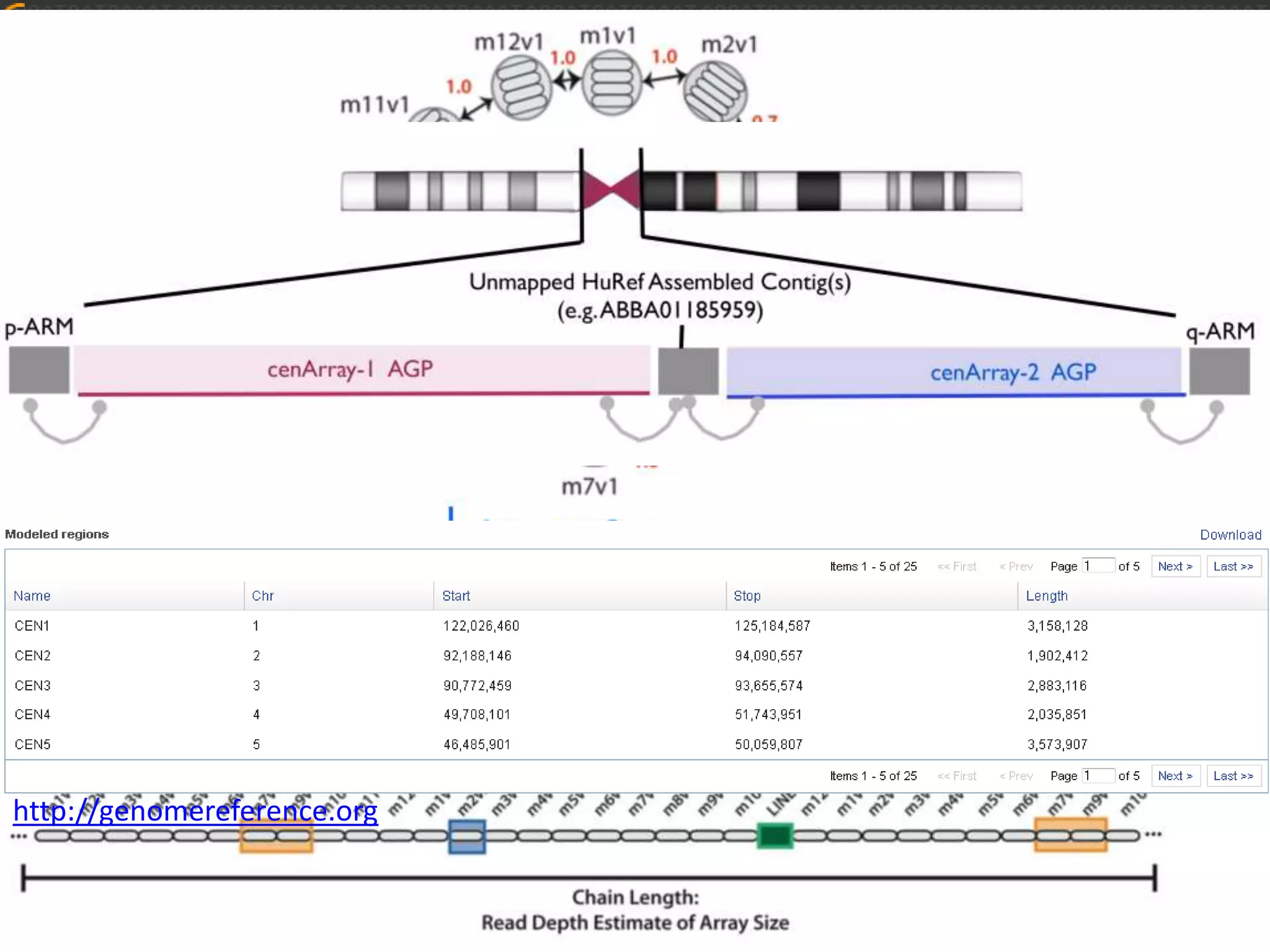
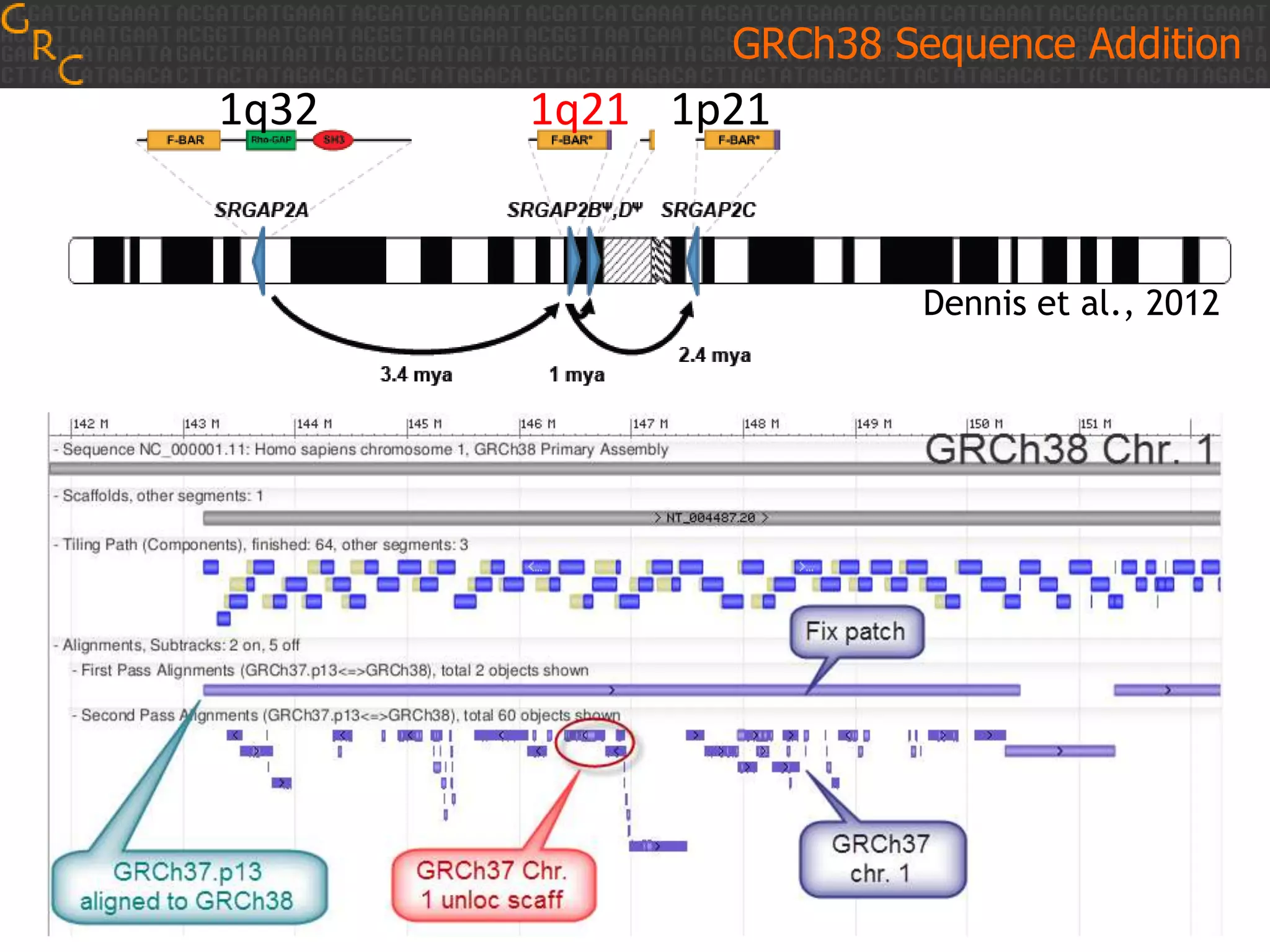
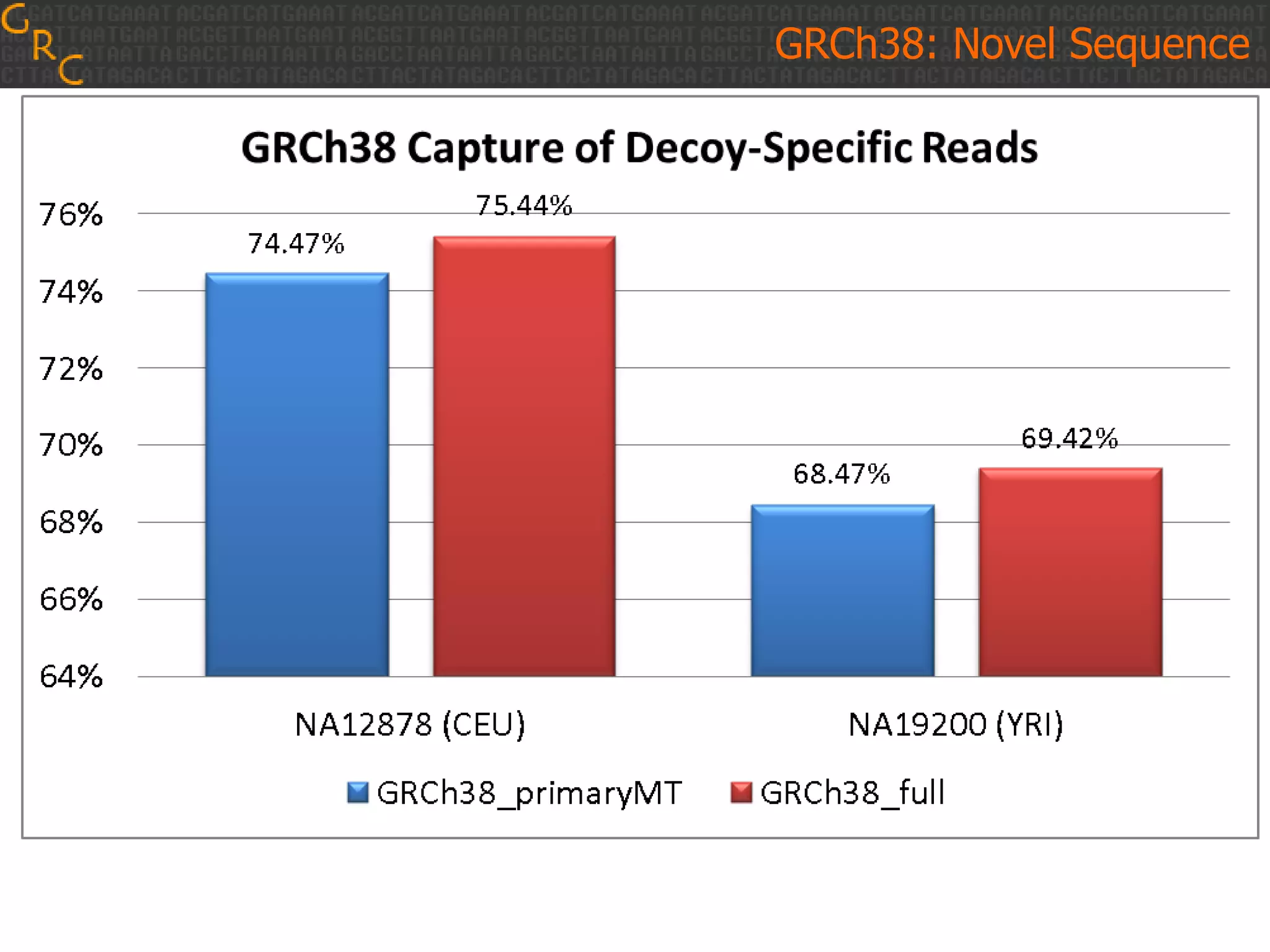
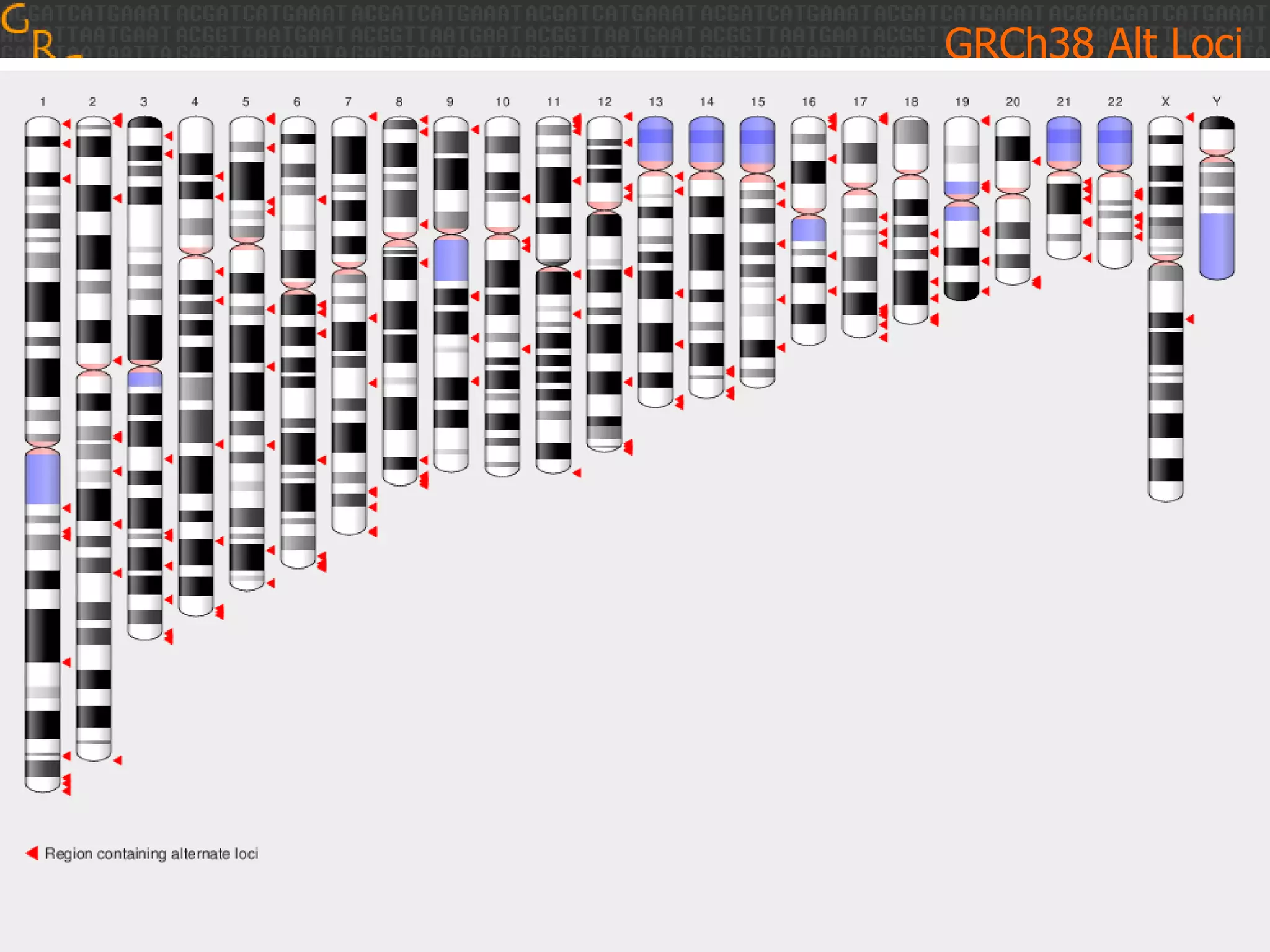
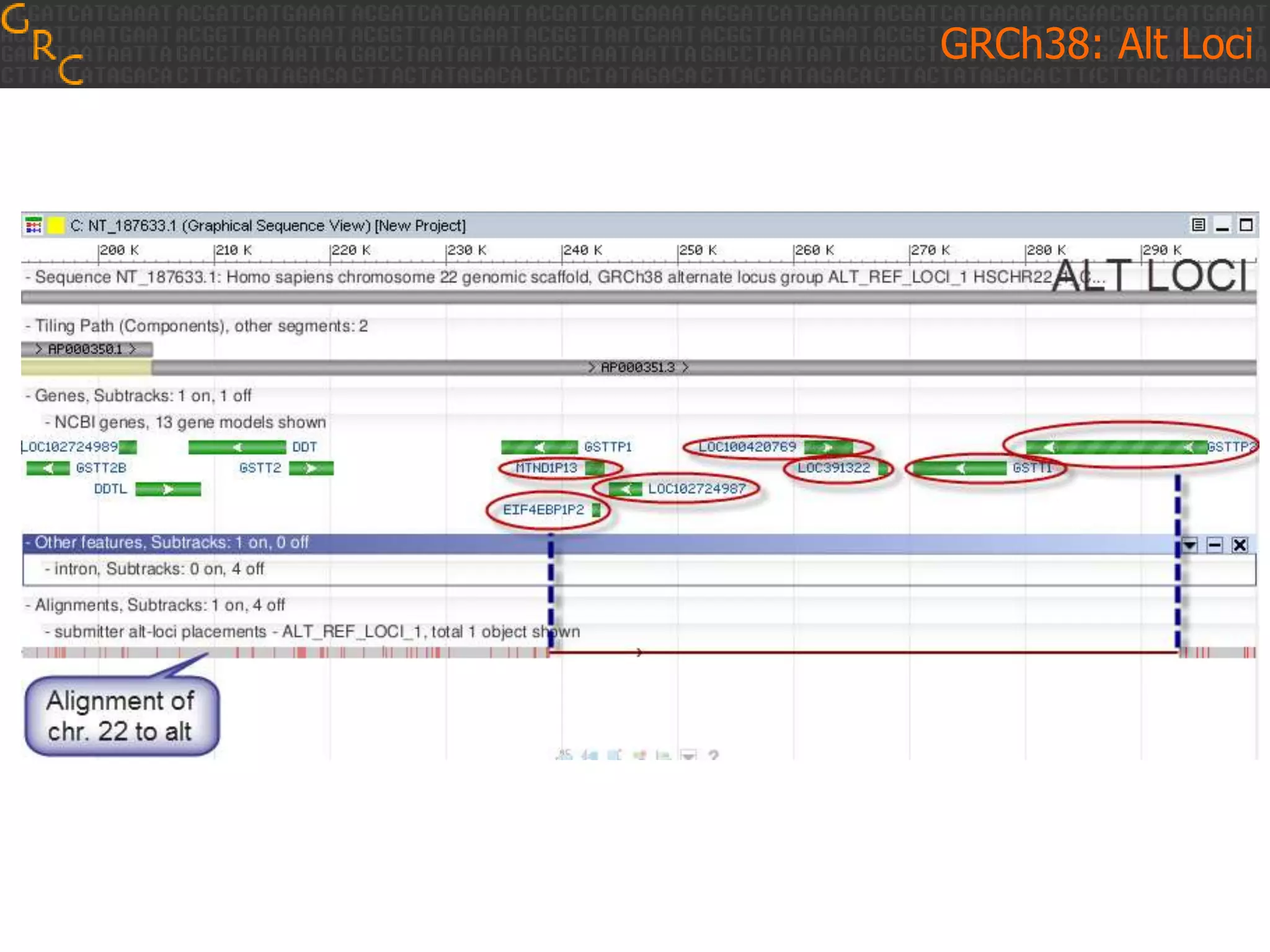
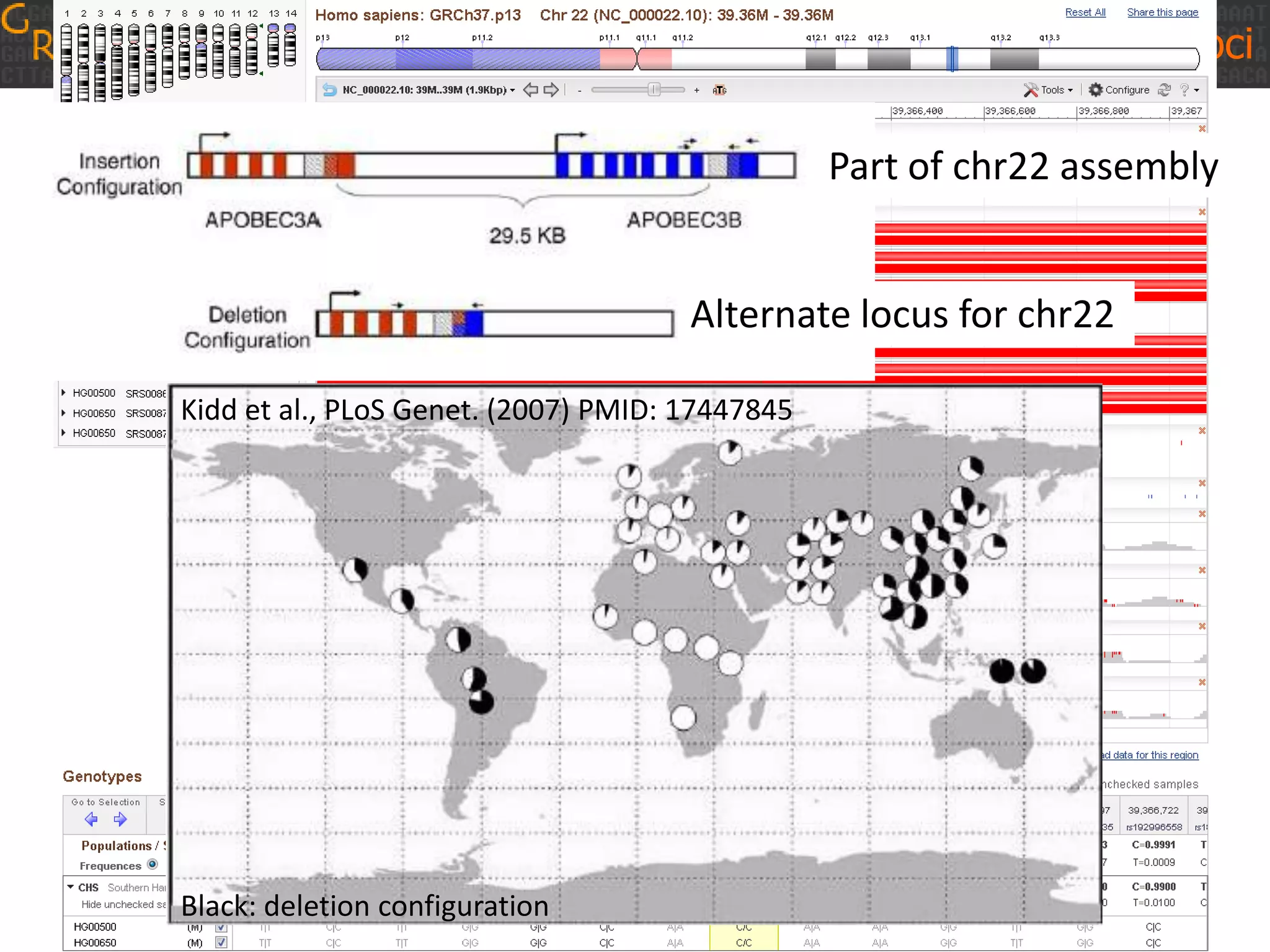
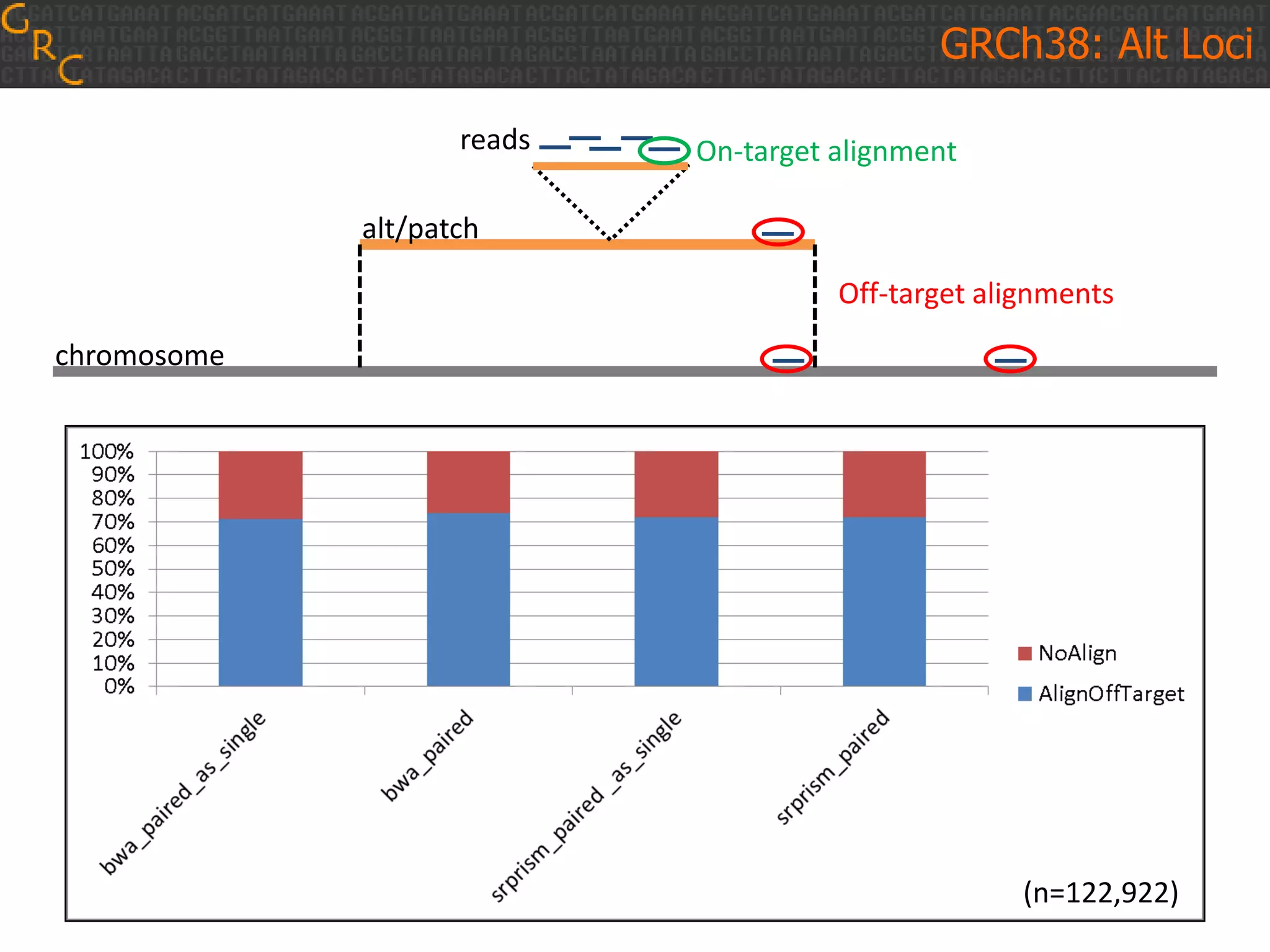
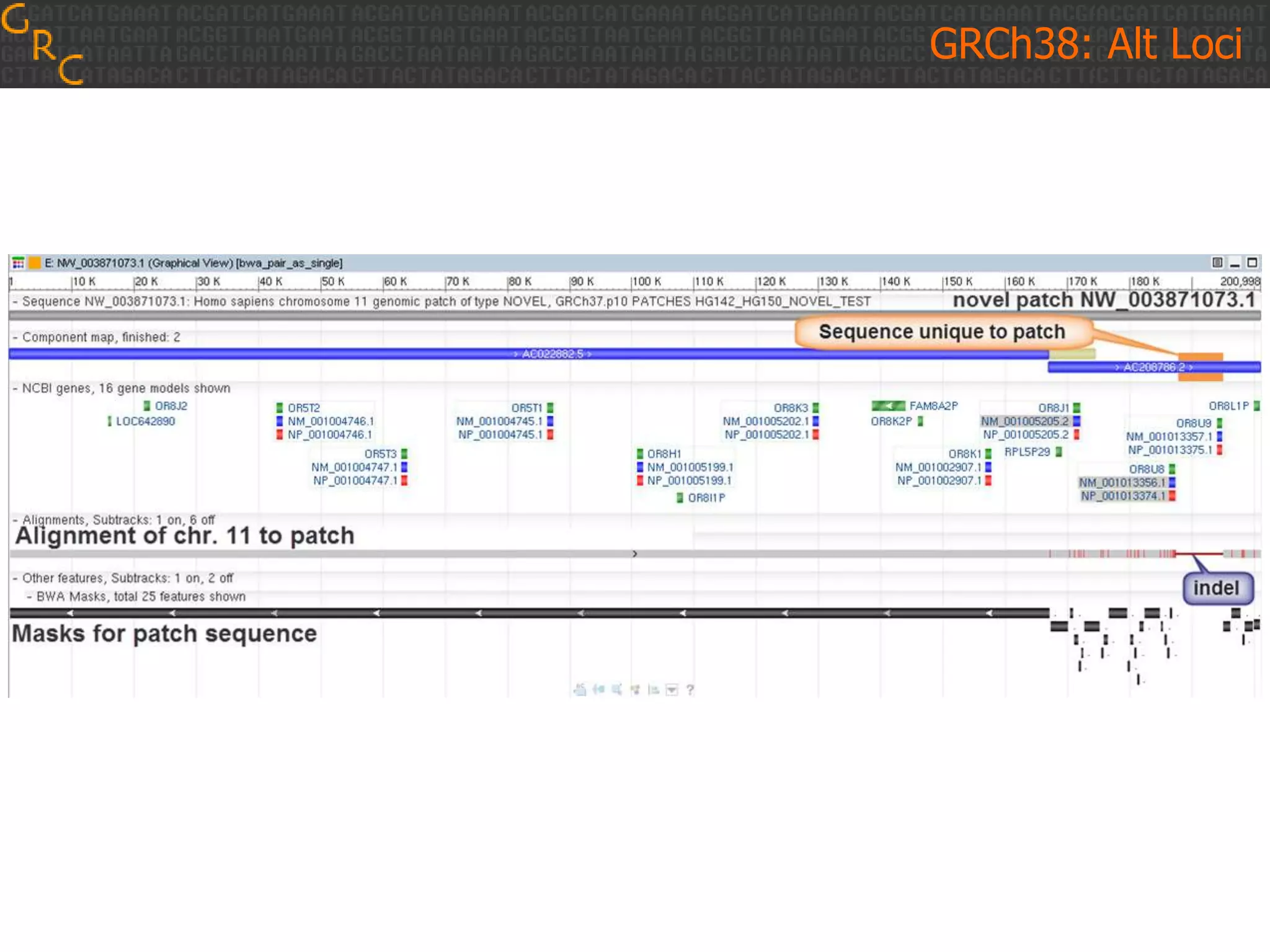
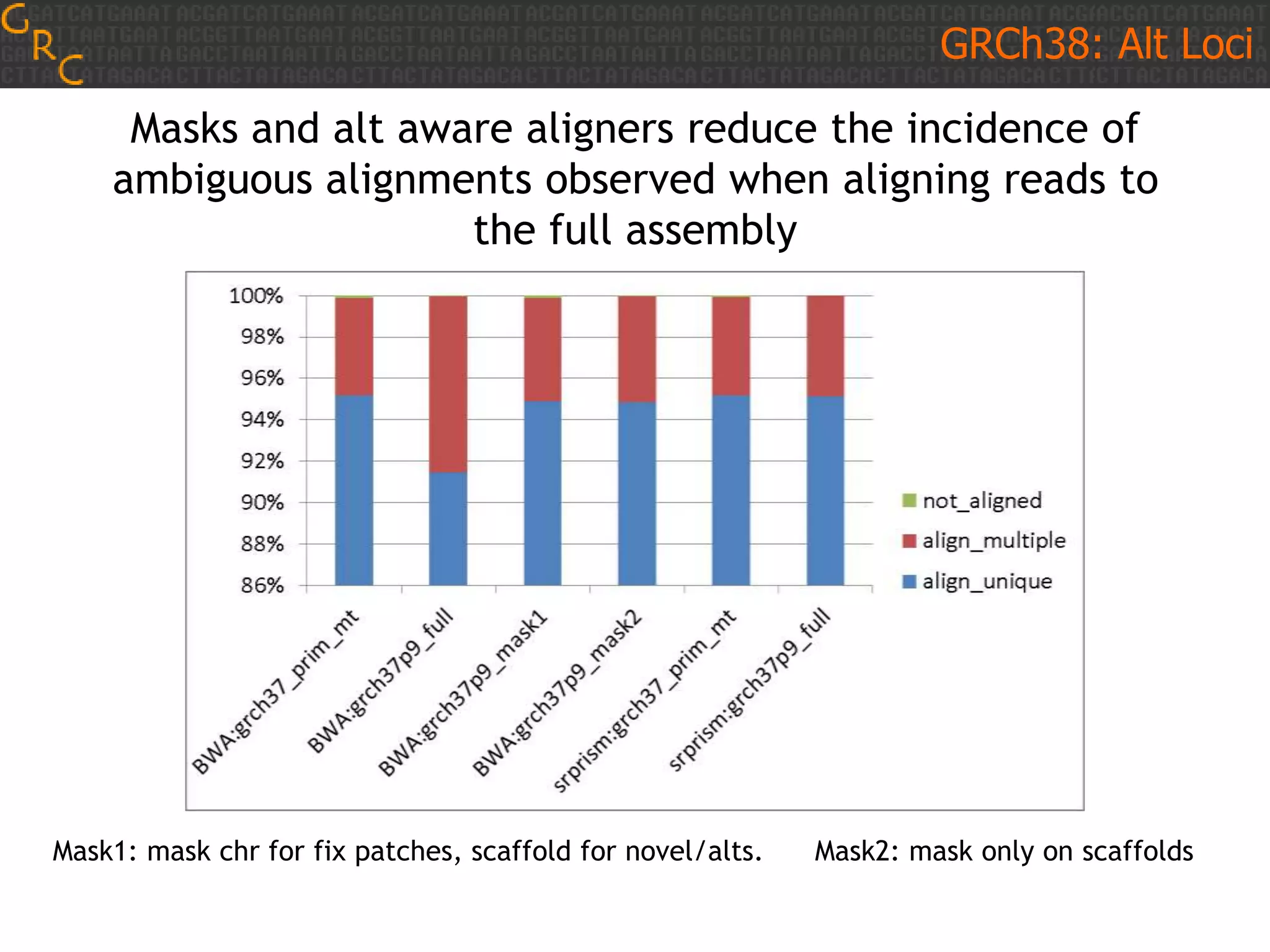
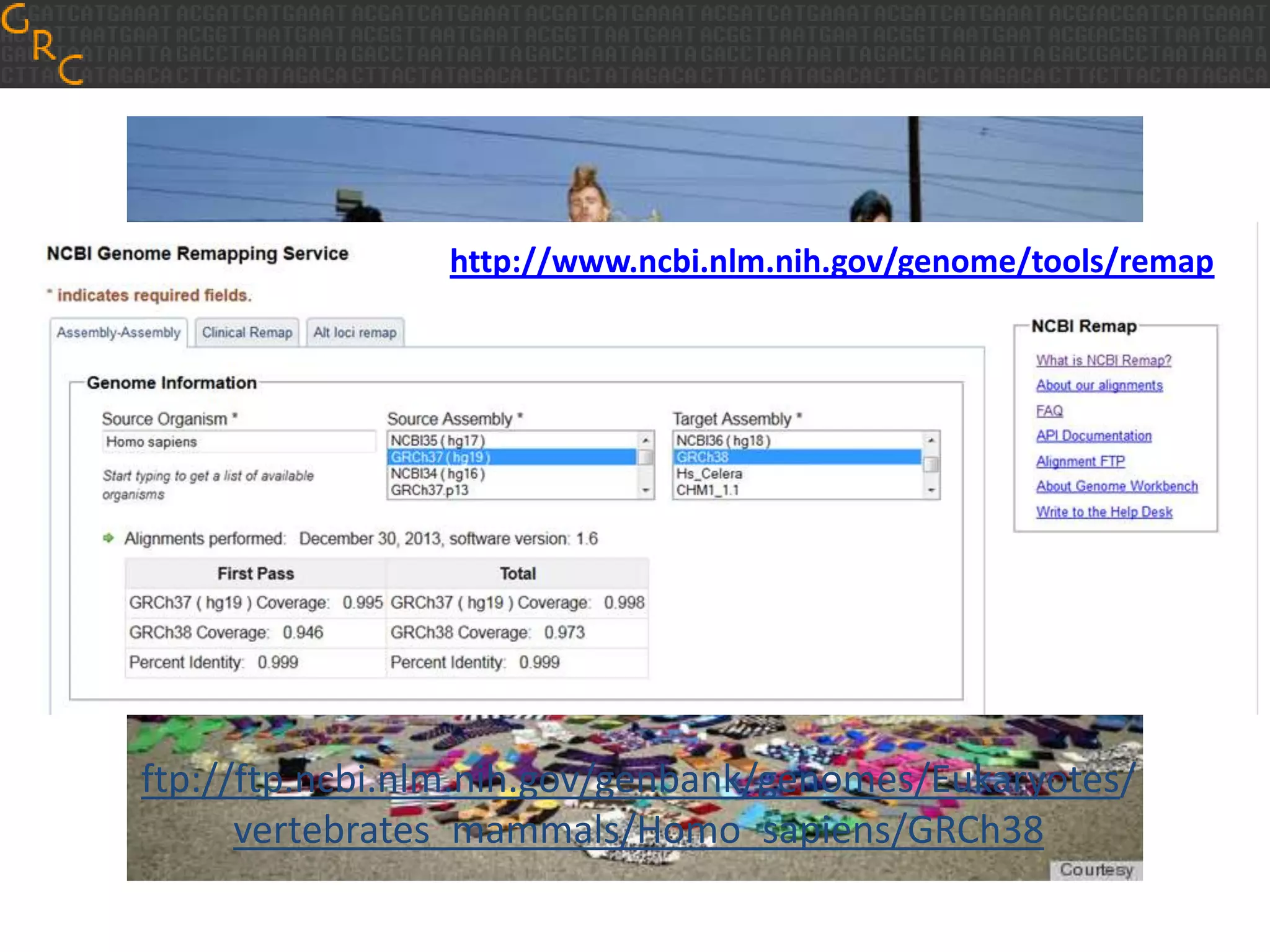
GRCh38 is a new version of the human reference genome that features several improvements over the previous version, GRCh37. It includes 178 regions comprising 3.15% of the genome sequence that have been updated based on new data. GRCh38 also includes 261 alternate loci comprising 3.6 Mb of novel sequence not present in GRCh37. Model centromeres have been added to chromosomes for the first time, representing heterochromatic regions totaling over 60 Mb. In addition, over 800 kb of novel sequence has been added through 73 patches of previously unrepresented DNA.














































![Coded Agents – with UiPath SDK + LangGraph [Virtual Hands-on Workshop]](https://cdn.slidesharecdn.com/ss_thumbnails/codedagentsdeck-251215155422-5497c599-thumbnail.jpg?width=640&height=640&fit=bounds)



