Download as PDF, PPTX










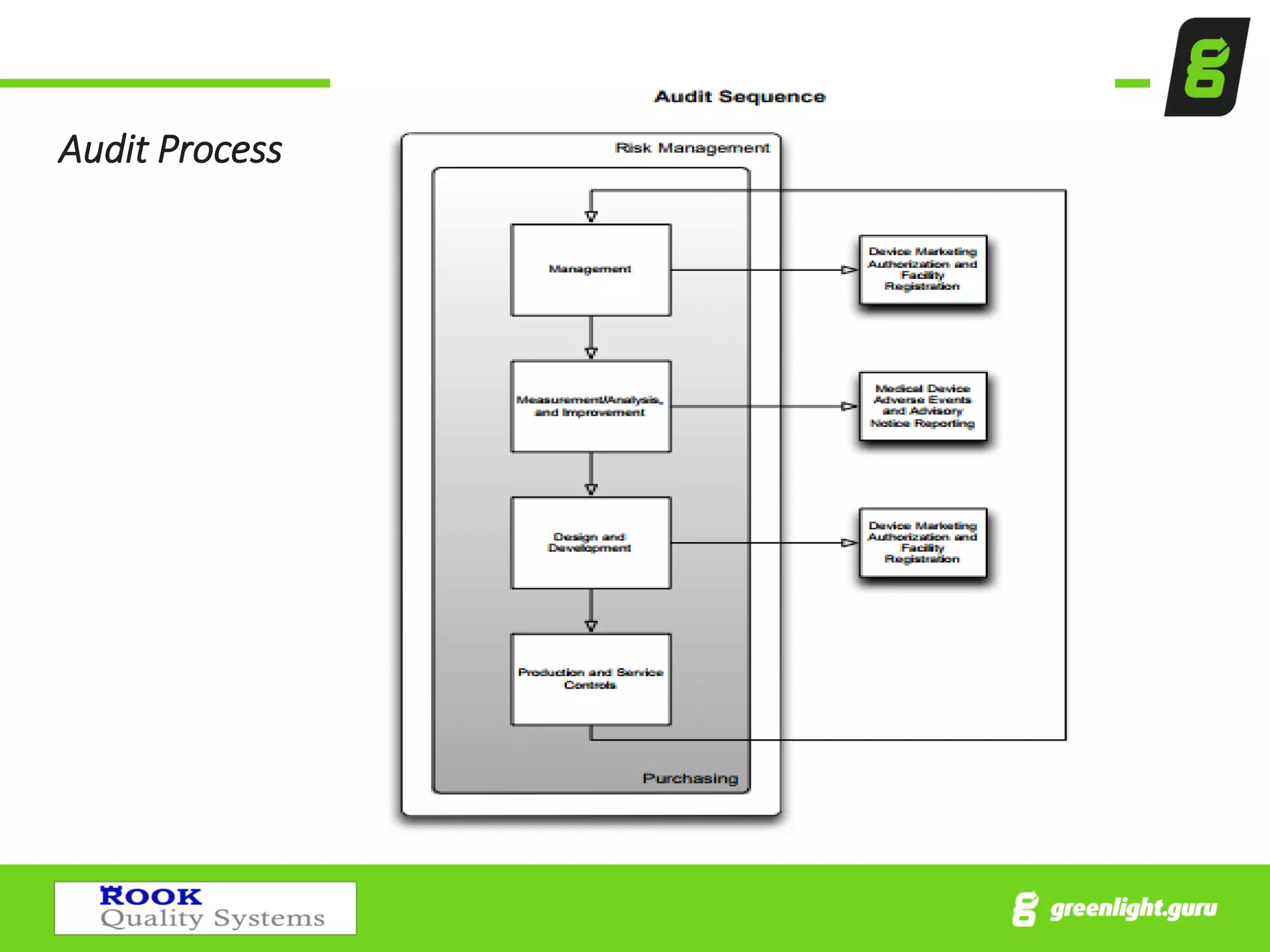


















The document provides an overview of the Medical Device Single Audit Program (MDSAP), which allows manufacturers to consolidate regulatory audits from multiple countries, including the USA, Canada, and Australia, into a single audit to streamline the approval process and reduce costs. It outlines the audit process, transition year changes related to ISO 13485:2016, and the regulatory impacts in different regions. It also emphasizes the importance of proper quality management systems and offers consulting services to assist companies in preparing for MDSAP audits.























































































