Download as PDF, PPTX



![MISSION
IMDRF/MC/N1FINAL:2014
“The mission of the IMDRF [International Medical Device
Regulators Forum] is to strategically accelerate international
medical device regulatory convergence to promote an efficient
and effective regulatory model for medical devices that is
responsive to emerging challenges in the sector while
protecting and maximizing public health and safety.”](https://image.slidesharecdn.com/339a871b-ec2f-4dee-868a-60debcbe0bf5-160116132517/75/MDSAP-4-2048.jpg)






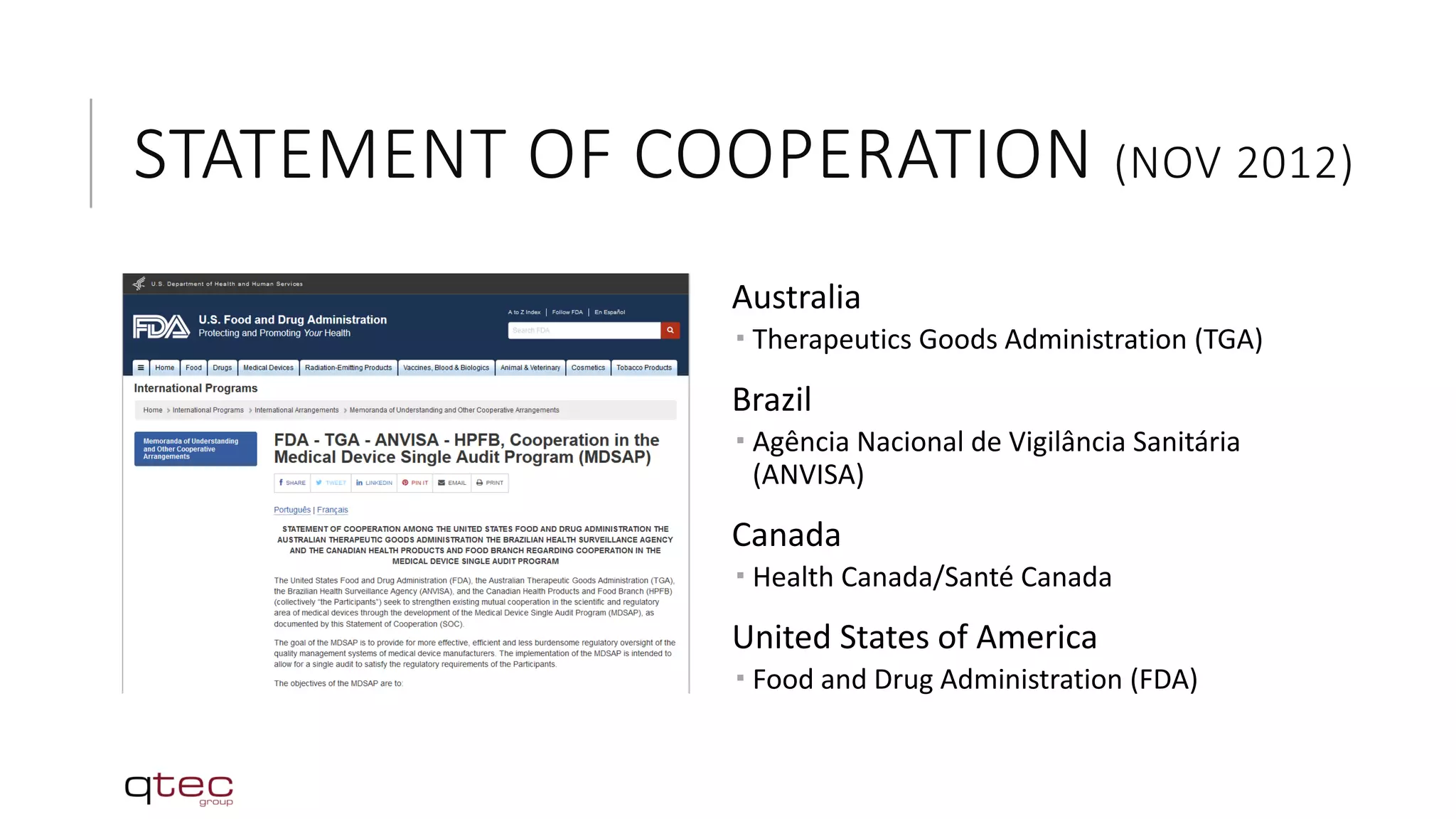







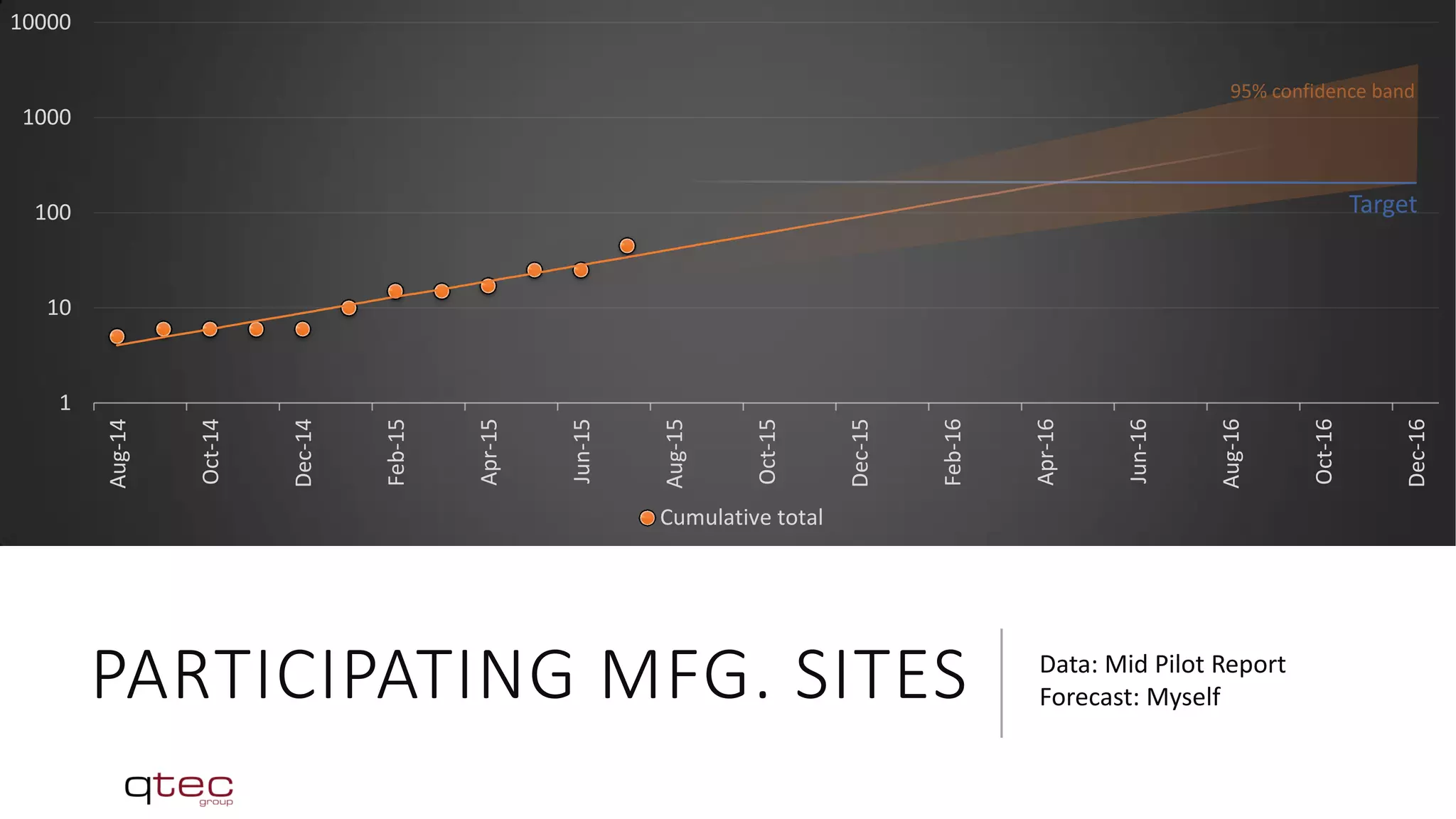













The Medical Device Single Audit Program (MDSAP) is an initiative by the International Medical Device Regulators Forum (IMDRF) to develop a harmonized audit program that allows medical device manufacturers to undergo a single regulatory audit to satisfy the requirements of multiple regulatory jurisdictions. The MDSAP pilot program began in 2014 and involves regulators from the US, Canada, Brazil, Australia and Japan. It aims to recognize third-party auditing organizations to conduct audits of medical device manufacturers according to a standardized audit process, with the goal of facilitating medical device trade while ensuring public health and safety.






































![Educo Life Science [gathering clinical evidence] [module 1]](https://cdn.slidesharecdn.com/ss_thumbnails/educo-gatheringclinicalevidence-module1-220128131137-thumbnail.jpg?width=640&height=640&fit=bounds)








