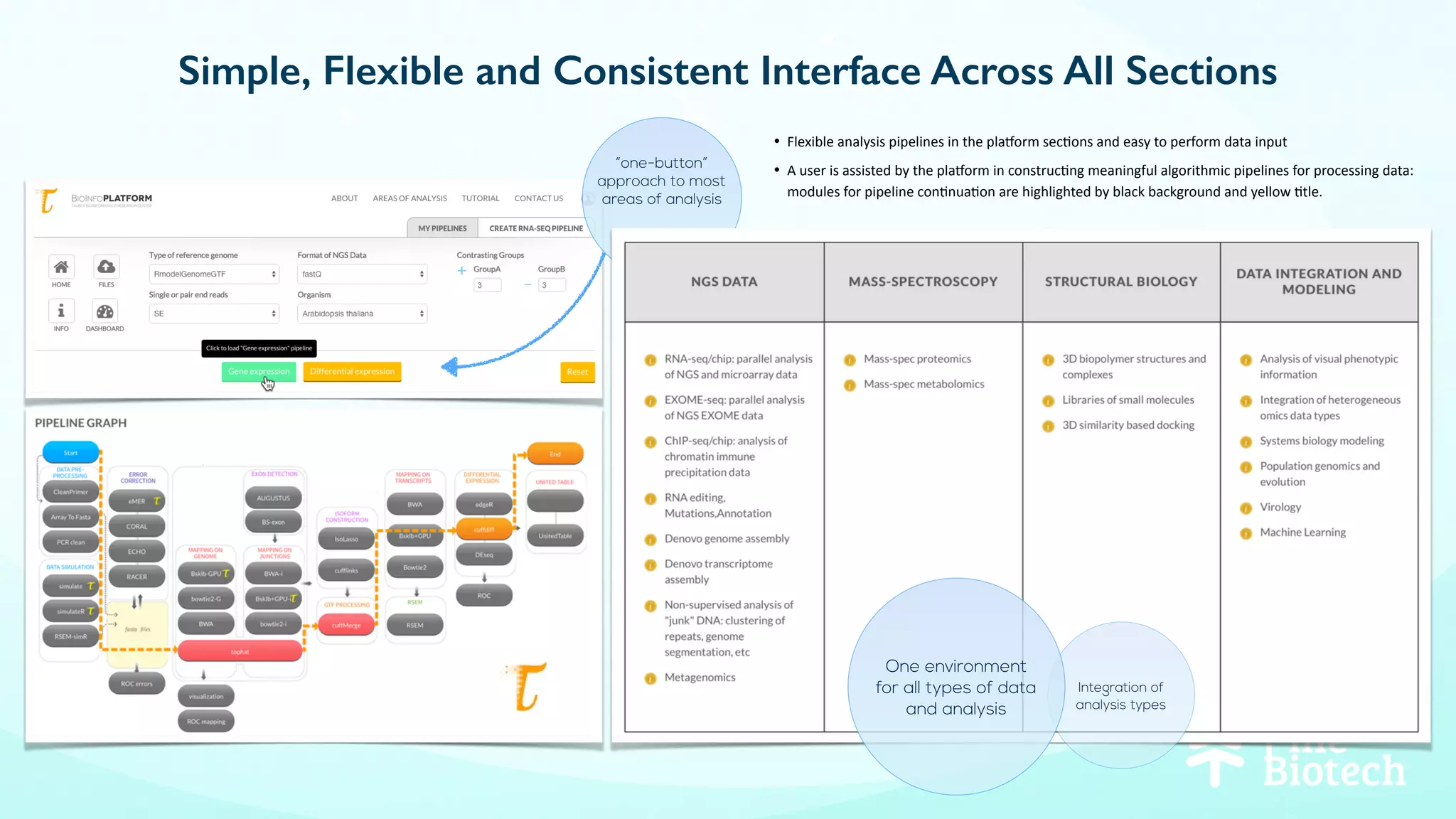
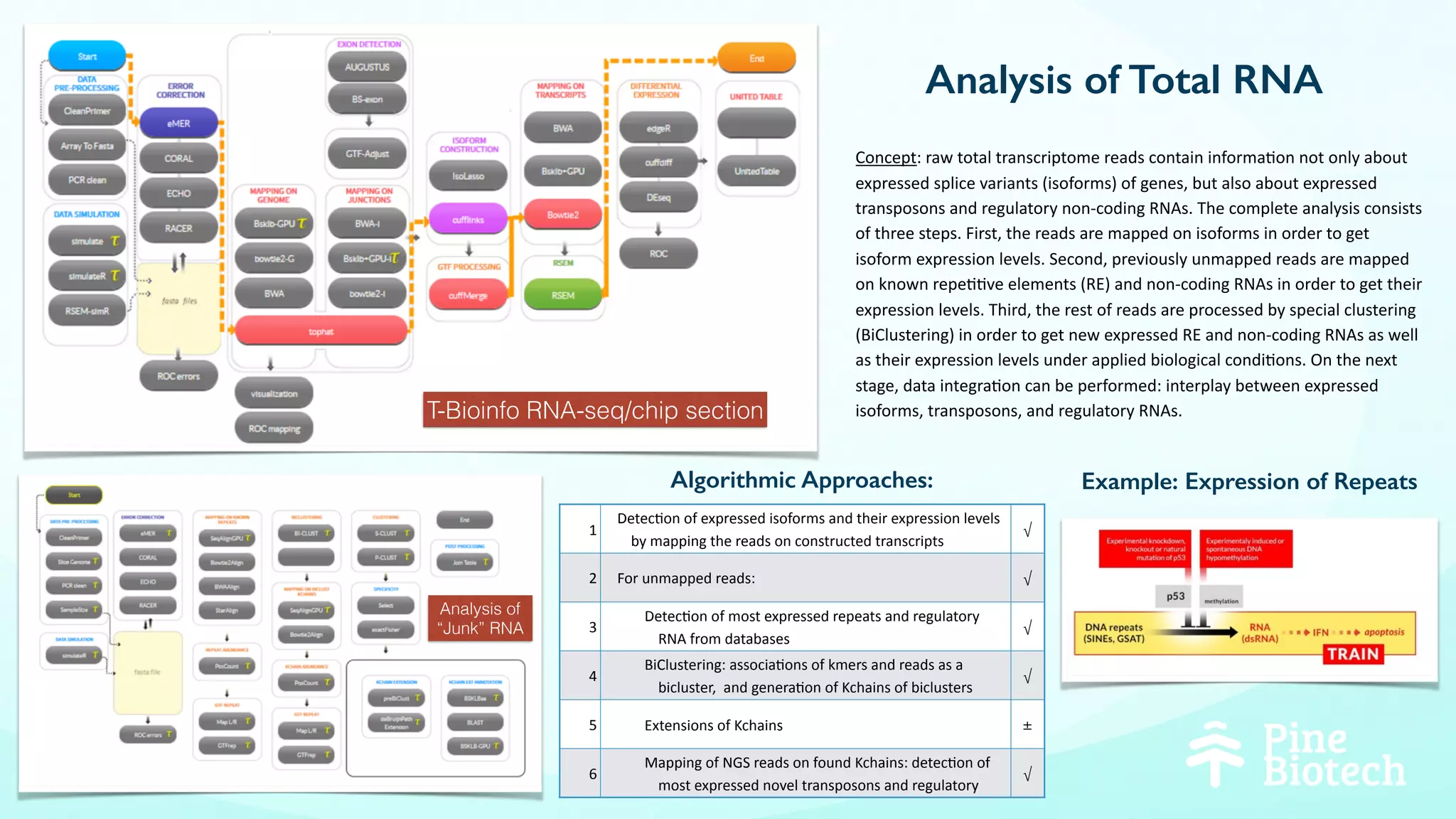
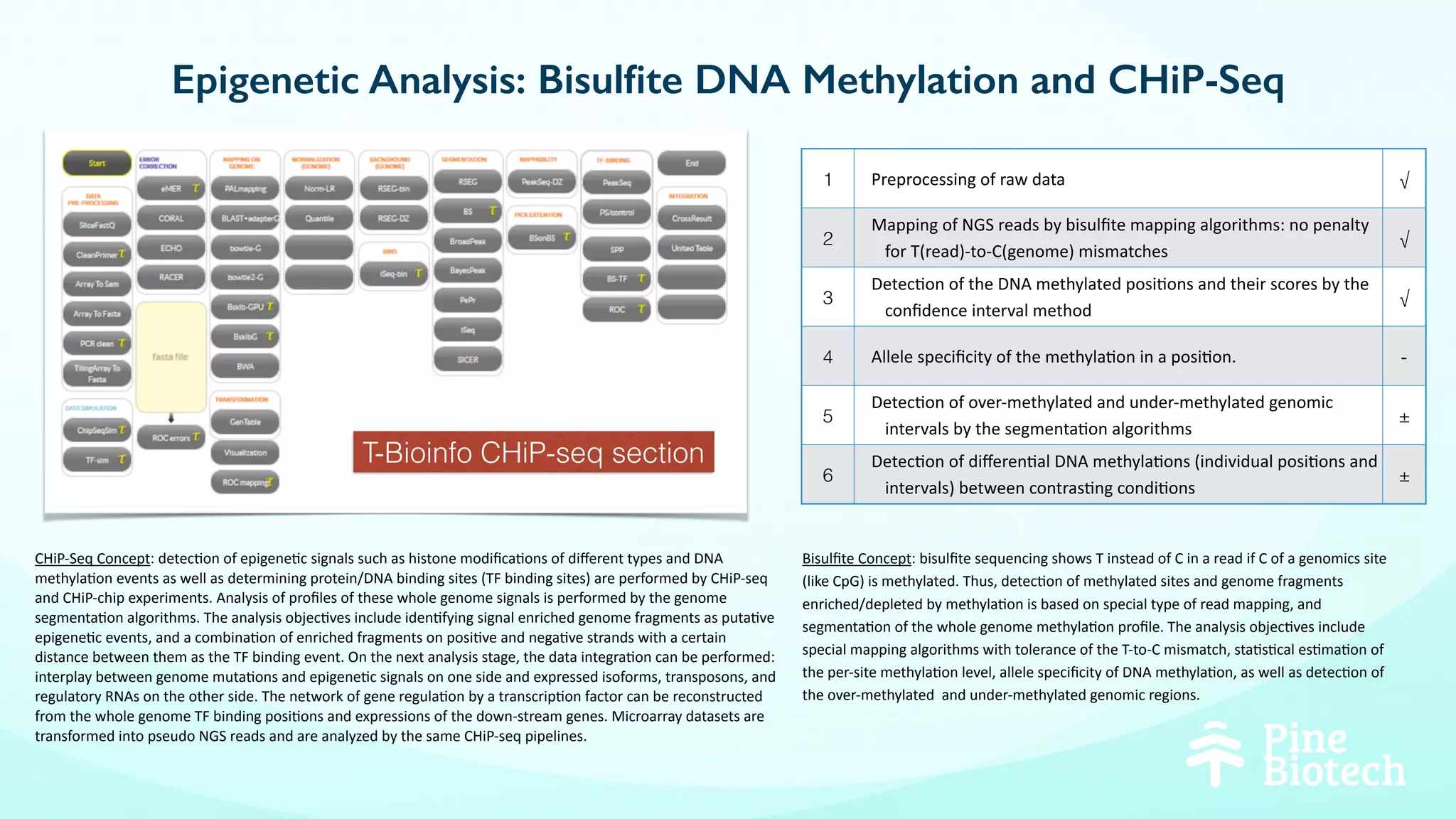
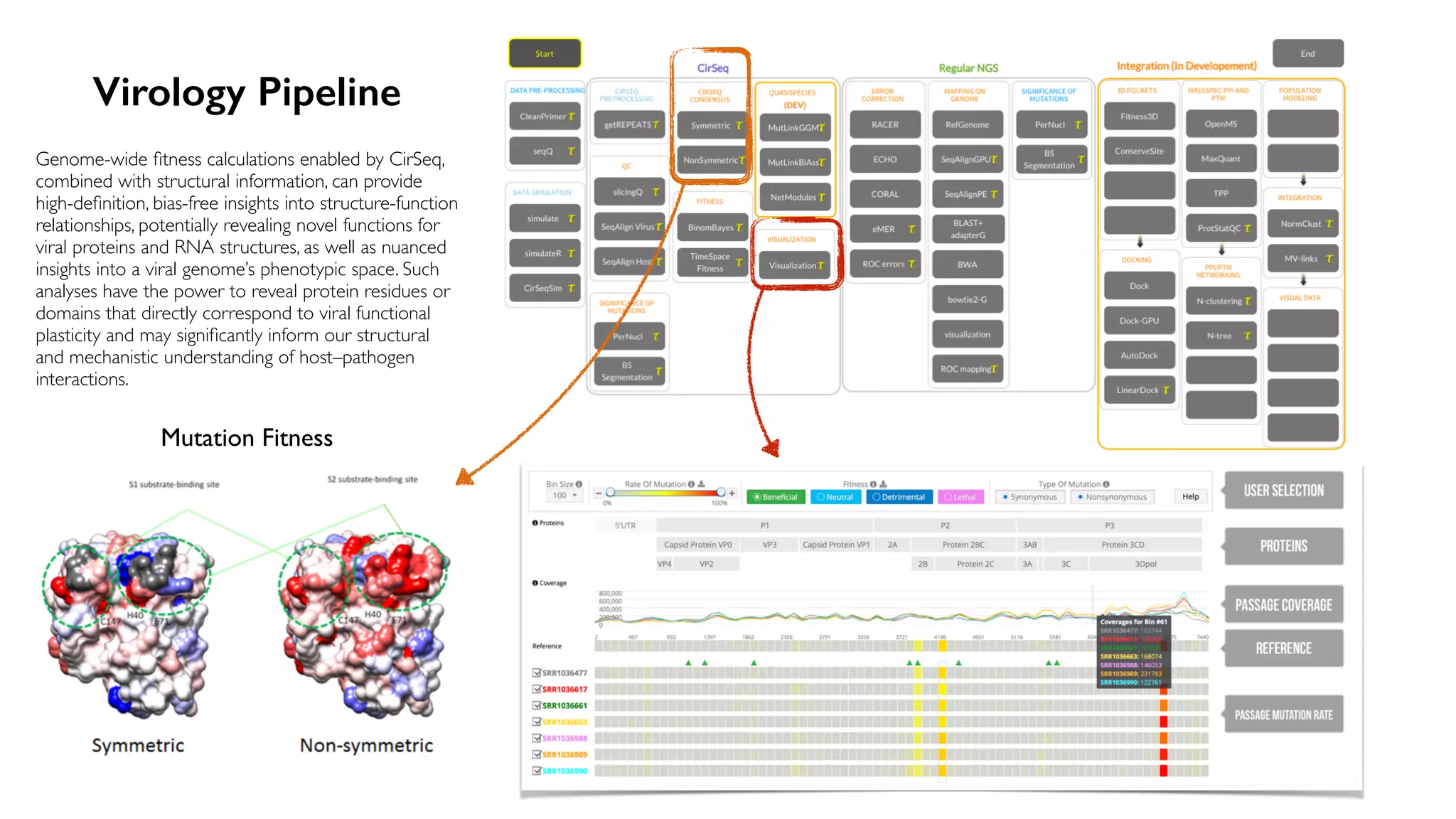
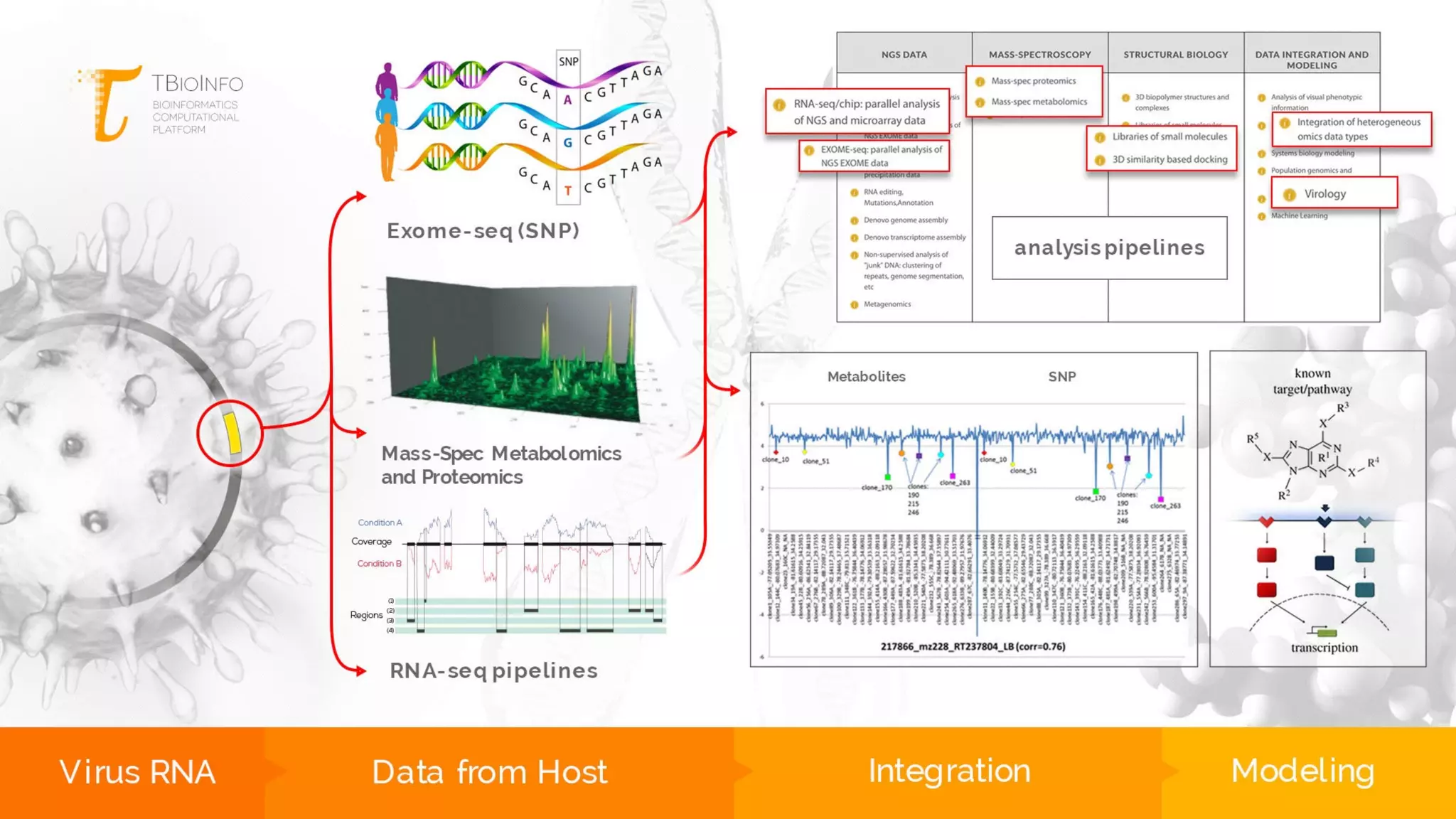
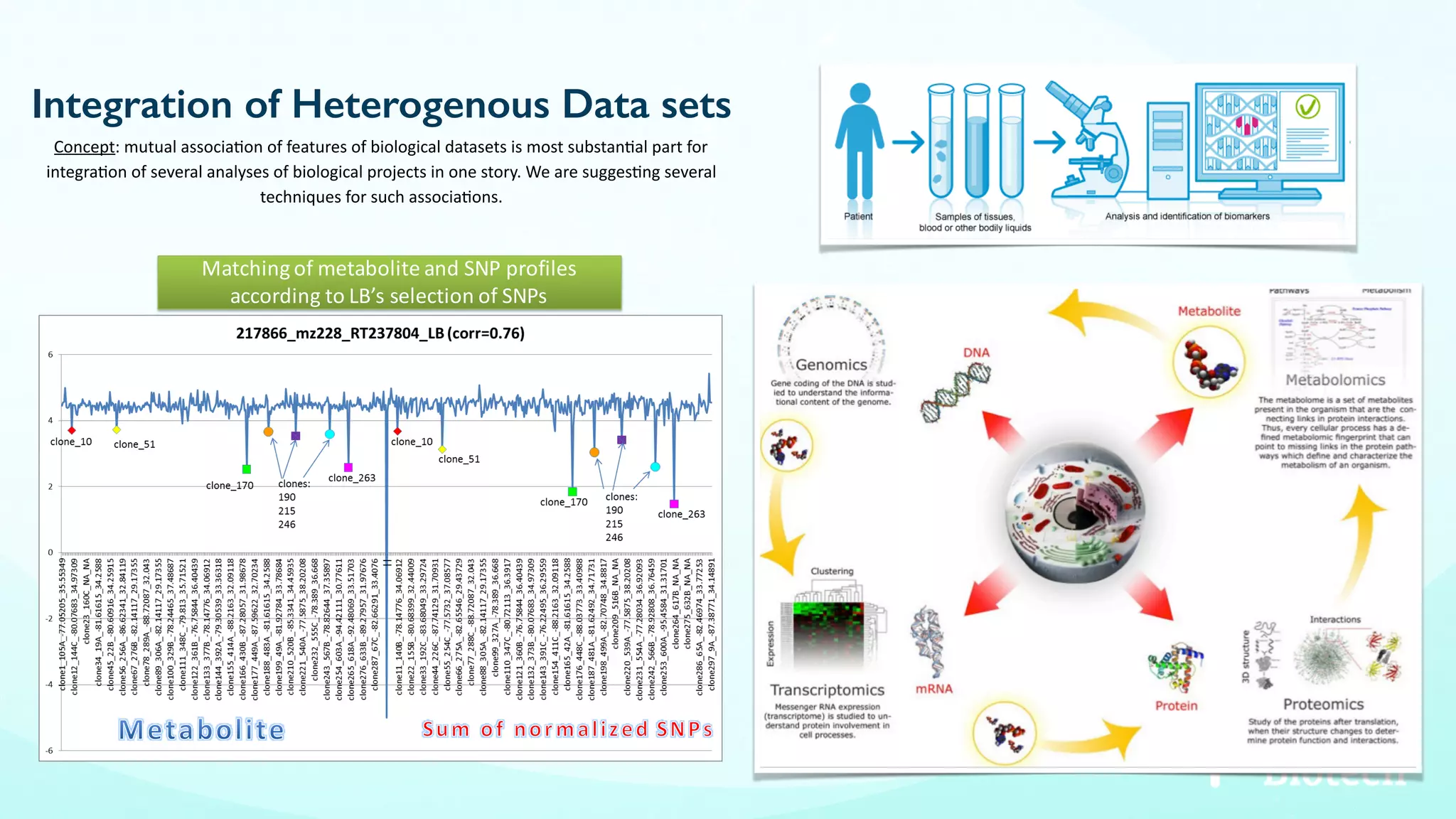
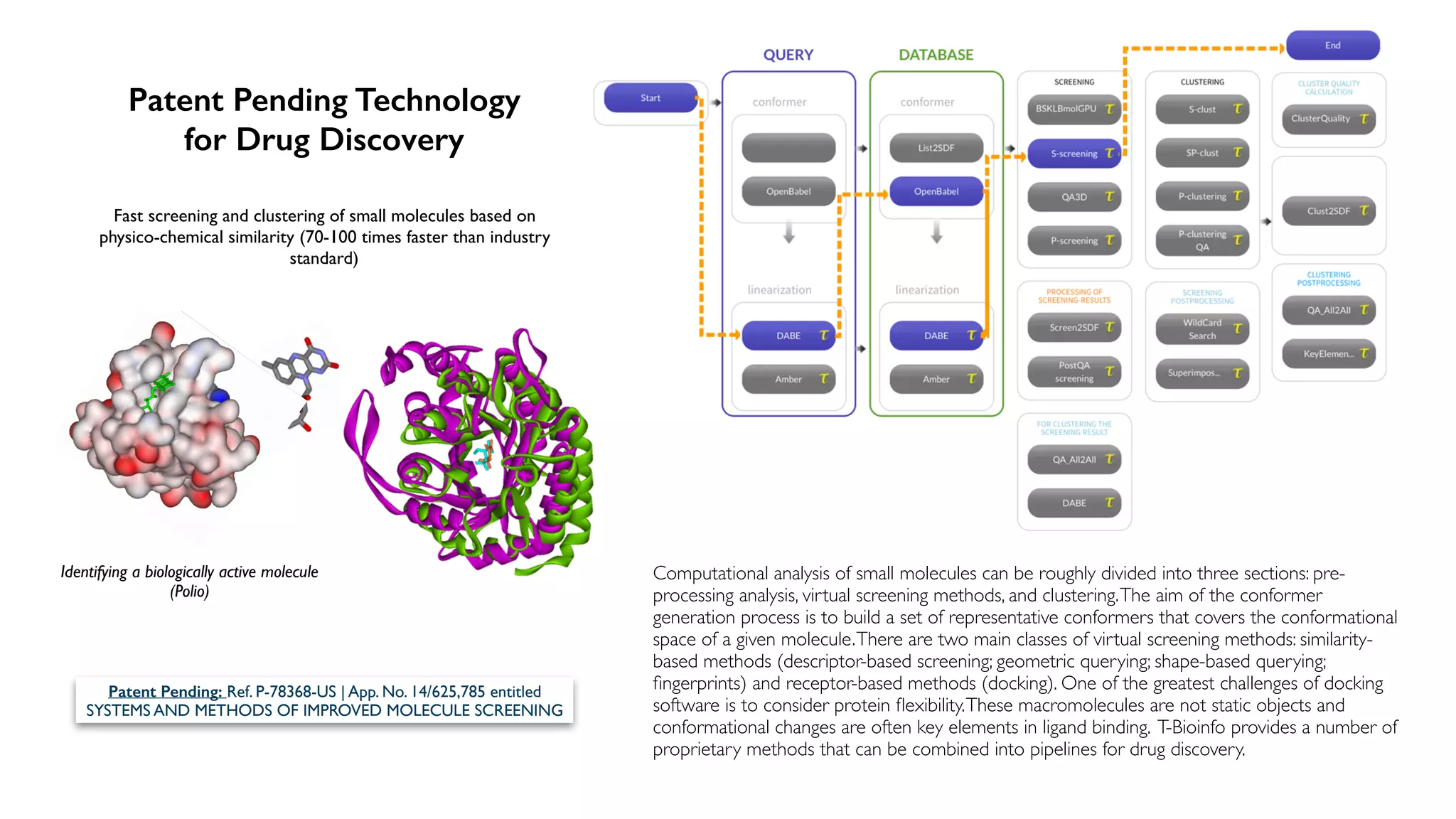
The document discusses the challenges and workflows of various mass-use pipelines in bioinformatics, particularly focusing on next-generation sequencing (NGS) analysis, epigenetics, and machine learning applications. It introduces t-bioinfo, a user-friendly computational platform designed for the integration and analysis of diverse biological data types, and outlines innovative workflows for RNA-seq, epigenetic analysis, and virology pipelines. Additionally, it highlights collaborations and commercialization efforts with research centers for drug discovery and analysis of small molecules.