Downloaded 10 times









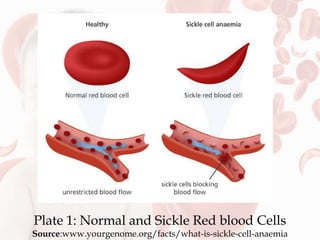

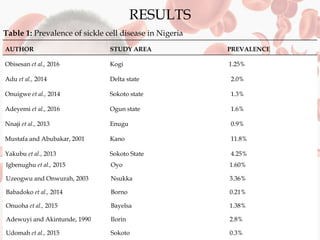
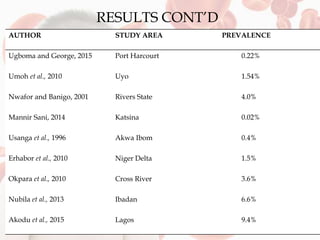
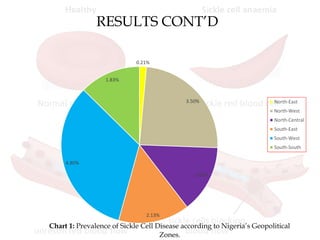
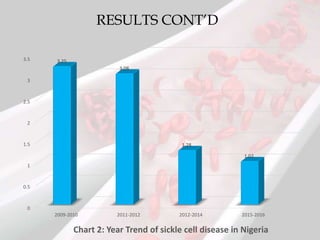



This document reviews sickle cell disease and its public health significance. It discusses how sickle cell disease results from an inherited abnormality in hemoglobin that causes red blood cells to take on a rigid, sickle shape, blocking blood flow and causing pain. The effects of sickle cell disease include frequent pain crises, psychological impacts, and reduced quality of life. While it places a burden on individuals and families, sickle cell trait provides some protection against malaria. The document also summarizes prevalence data on sickle cell disease in Nigeria and trends over time. It concludes that sickle cell disease remains a significant health issue in Nigeria and recommends improving awareness, screening programs, and affordable treatment technologies.



































































