WHAT IS SEQUENCEALIGNMENT?
In bioinformatics, a sequence alignment is
a way of arranging the sequences
of DNA, RNA, or protein to identify
regions of similarity that may be a
consequence of functional, structural,
or evolutionary relationships between the
sequences.
Aligned sequences
of nucleotide or amino acid residues are
typically represented as rows within
a matrix.
Gaps are inserted between the residues so
that identical or similar characters are
aligned in successive columns.
3.
A sequence alignment,produced by ClustalO, of mammalian histone proteins. Sequences are the amino
acids for residues 120-180 of the proteins. Residues that are conserved across all sequences are highlighted in
grey. Below the protein sequences is a key denoting conserved sequence (*), conservative mutations (:), semi-
conservative mutations (.), and non-conservative mutations ( ).
4.
IMPORTANCE OF SEQUENCEALIGNMENT
Sequence determines structure and structure determines function.
By studying sequence similarities, we can find the correlation between sequences,
structures, functions and evolutionary linkages.
It is important for newly determined sequence to compare it with other sequences that
already exist in the databases, to determine the structure, functions and evolutionary
linkages of newly determined sequence with existing sequences in the databases.
Thus the process of comparison is sequence alignment.
The process in which sequences are compared by searching for common character patterns
and establishing residue to residue correspondence among related sequences is called
sequence alignment.
5.
INTERPRETATION
If two sequencesin an alignment share a common ancestor, mismatches can
be interpreted as point mutations and gaps as indels (that is, insertion or
deletion mutations) introduced in one or both lineages in the time since they
diverged from one another.
In sequence alignments of proteins, the degree of similarity between amino
acids occupying a particular position in the sequence can be interpreted as a
rough measure of how conserved a particular region or sequence motif is
among lineages.
The absence of substitutions, or the presence of only very conservative
substitutions (that is, the substitution of amino acids whose side chains have
similar biochemical properties) in a particular region of the sequence, suggest
that this region has structural or functional importance.
6.
WHAT IS INDEL?
Indelis a molecular biology term for the insertion or the deletion of bases in the DNA of
an organism.
It has slightly different definitions between its use in evolutionary studies and its use in
germ-line and somatic mutation studies.
In evolutionary studies, indel is used to mean an insertion or a deletion and indels simply
refers to the mutation class that includes both insertions, deletions, and the combination
thereof, including insertion and deletion events that may be separated by many years,
and may not be related to each other in any way.
In germline and somatic mutation studies, indel describes a special mutation class,
defined as a mutation resulting in both an insertion of nucleotides and a deletion of
nucleotides which results in a net change in the total number of nucleotides, where both
changes are nearby on the DNA.
A microindel is defined as an indel that results in a net change of 1 to 50 nucleotides.
7.
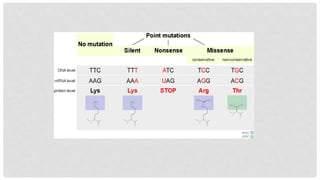
WHAT ARE POINTMUTATIONS?
A point mutation, or single base modification, is a type of mutation that causes a
single nucleotide base substitution, insertion, or deletion of the genetic material,
DNA or RNA. The term frameshift mutation indicates the addition or deletion of a
base pair.
9.
Nonsense mutations
Codefor a stop, which can truncate the protein.
A nonsense mutation converts an amino acid codon
into a termination codon.
This causes the protein to be shortened because of
the stop codon interrupting its normal code.
How much of the protein is lost determines whether
or not the protein is still functional.
Missense mutations
Code for a different amino acid.
A missense mutation changes a codon so that a
different protein is created, a non-synonymous
change.
Conservative mutations
Result in an amino acid change.
However, the properties of the amino acid remain the
same (e.g., hydrophobic, hydrophilic, etc.).
At times, a change to one amino acid in the protein is not
detrimental to the organism as a whole.
Most proteins can withstand one or two point mutations
before their function changes.
Non-conservative mutations
Result in an amino acid change that has different
properties than the wild type.
The protein may lose its function, which can result in a
disease in the organism.
For example, sickle-cell disease is caused by a single
point mutation (a missense mutation) in the beta-
hemoglobin gene that converts a GAG codon into GUG,
which encodes the amino acid valine rather than glutamic
acid.
The protein may also exhibit a "gain of function" or
become activated, such is the case with the mutation
changing a valine to glutamic acid in the braf gene; this
leads to an activation of the RAF protein which causes
unlimited proliferative signalling in cancer cells.
These are both examples of a non-conservative (missense)
mutation.
10.
Silent mutations
Code forthe same amino acid.
A silent mutation has no effect on the functioning of the protein.
A single nucleotide can change, but the new codon specifies the same amino
acid, resulting in an unmutated protein.
This type of change is called synonymous change, since the old and new codon
code for the same amino acid.
This is possible because 64 codons specify only 20 amino acids.
Different codons can lead to differential protein expression levels, however.
11.
DNA and proteinare the products of evolution.
DNA and protein are biological macromolecules composed of nucleotides and amino acid
to form linear sequences and these sequences determine the primary structure of the
molecules.
These molecules also store the history of evolution.
The presence of evolutionary traces in the sequences because some of the residues that
perform key functional and structural roles tend to be preserved by natural selection.
While other residues that are less common tend to mutate frequently. For example active
site residues of an enzyme family tend to be conserved because they are responsible for
catalytic functions.
Hence, by alignment, we can identify the conserved and varied region (patterns of
conservation and variations).
12.
The degree ofsequence conservation in the alignment demonstrate the evolutionary
relatedness among different specie while variations between sequences demonstrate
the changes that have occurred during evolution in the form of substitution,
insertion and deletion.
We can demonstrate the function of unknown sequences by identifying the
evolutionary relationship between sequences.
If we find a “significant similarity” among sequences, so we can say that they
belong to same family (protein family).
By using the information (structure and function) of known protein sequence, we
can predict the structure and function of uncharacterized sequences.
If two sequences have significant similarity, so we can say that they have been
from common ancestor.
13.
SIGNIFICANCE OF SEQUENCEALIGNMENT
It is helpful in the determination of,
Function
Structure
Evolutionary relationship
14.
METHODS OF SEQUENCEALIGNMENT
Two methods
Pair wise sequence alignment
Multiple sequence alignment
PAIR WISE SEQUENCEALIGNMENT
It is the process of comparison of two sequences.
Alignment has three aspects,
Quantity To what degree sequences are similar (%)
Quality Regions of similarity in a given sequence.
Optimal alignment The maximum similarity and the least differences.
17.
We can comparediseased genome/proteome to healthy one. We also compare
unknown gene with known gene. We can compare sequence of one organism with
other organisms that how closely they are related. We can also compare two
proteins of same family.
In pairwise sequence alignment, homology, similarity and identity are important.
18.
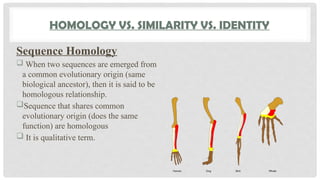
HOMOLOGY VS. SIMILARITYVS. IDENTITY
Sequence Homology
When two sequences are emerged from
a common evolutionary origin (same
biological ancestor), then it is said to be
homologous relationship.
Sequence that shares common
evolutionary origin (does the same
function) are homologous
It is qualitative term.
19.
Sequence Similarity
It isthe percentage of align residues that are similar in physiochemical
properties like size, charge, hydrophobicity. It is quantitative term
For example, subtilisin and chymotrypsin are homologous but their structure
and size are different.
ATCGGC and ATCGCG are similar
Similarity is based on physiochemical and biochemical properties of amino
acid that how close they are?
Similarity is actually the same nature of two amino acids
Similarity refers to the percentage of aligned residues that have similar
physiochemical properties.
20.
SEQUENCE IDENTITY
It refersto the percentage of matches of same amino acids residues between two
aligned sequences.
ATCGGC and ATCGGC are Identical
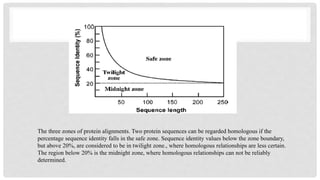
Safe zone: If two protein sequences are 30% or more than 30% identical, so they
are in safe zone means they are homologous and closely related to same
ancestors, such zone is safe zone.
21.
Twilight zone: Whentwo sequences have less than 25% identity.
Identity falls between 20% is basically in twilight zone.
So they are whether homologous or non homologous. This range of identity is called
twilight zone.
Homologous ----- similarity from same ancestor
Non homologous---- similarity from non similar ancestor.
Midnight zone: If identity of two sequences is less than 20% so unrelated sequences
are present, they did not connect to same ancestor, such zone is called mid night
zone.
22.
The three zonesof protein alignments. Two protein sequences can be regarded homologous if the
percentage sequence identity falls in the safe zone. Sequence identity values below the zone boundary,
but above 20%, are considered to be in twilight zone., where homologous relationships are less certain.
The region below 20% is the midnight zone, where homologous relationships can not be reliably
determined.
23.
TYPES OF PAIRWISE SEQUENCE ALIGNMENT
Two types of pair wise sequence alignment,
Local alignment
Global alignment
24.
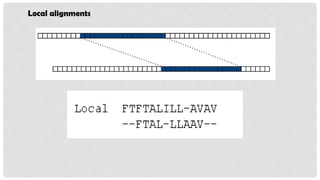
LOCAL ALIGNMENT
Localalignment only aligns the most similar regions between sequences.
Local alignments are more suitable for aligning sequences.
It determines the local regions of highest level of similarity/identity between
two sequences.
In local alignment, the alignment stops at the end of regions of identity.
Dashes in alignments indicate that these sequences are not included.
Local alignment is used to find,
Conserved domains
Conserved nucleotide pattern
Protein and DNA sequencing
GLOBAL ALIGNMENT
In thisalignment, entire sequence is aligned.
Two sequences are assumed to be similar over their entire sequence.
Sequences of high similarity and approx same length are suitable candidate for
global alignment.
Entire length of both same sequence length is aligned.
29.
SIGNIFICANCE OF PAIRWISE ALIGNMENT
We can,
Identify shared domain
Identify duplicated region
Identify important features like catalytic domains and disulphide bridges
Compare gene and its product.
30.
METHODS OF PAIRWISE SEQUENCE ALIGNMENT
Four methods
Align by hand
Dot plot
Dynammic programming (slow, optimal)
K- tuple word method ( FASTA and BLAST)
31.
ALIGN BY HAND
Ituses scoring system.
If the characters are identical, so positive score is given, if characters are different ,
so negative score to alignment is given ( usually called the quality).
32.
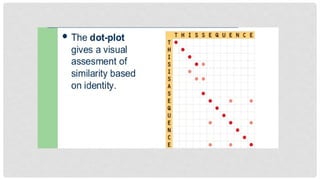
DOT PLOT
Also calleddot matrix method.
It is the graphical presentation of pairwise sequence alignment.
In this, two sequences are placed on graph against their axis. If the residue of
both sequences are matched so dot is placed and if not, so the place is left.
Afterwards diagonal is drawn that covers most of dots. If there is interruptions in
middle of diagonal, so it indicated the insertions or deletions.
Major problem in dot plot is the emergence of noises. These are small diagonals
that don’t provide significant or meaningful information.
34.
Advantages
Can be usedto align protein and nucleotide sequences.
Helpful to analyze long insertion, deletions and repetitions.
Provides pictorial statement of the relationship between two sequences.
Disadvantages
This method does not give perfect optimal alignment.
It is difficult for the methods to scale up multiple alignment.
Web tools for dot plot
Webserver, dot matcher, dottup, dot helix, matrix plot.
35.
DYNAMIC PROGRAMMING
It alsodetermines optimal alignment by matching pair between two sequences.
It creates two dimensional alignment grid (matrix) in which two sequences are
compared.
Identical match is assigned a score 1, mismatch 0 and gap penalty -1.
Gap is due to insertions/ deletions.
Time consuming procedure.
37.
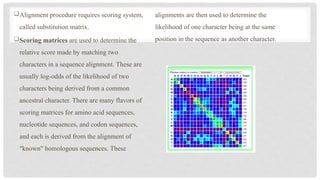
Alignment procedure requiresscoring system,
called substitution matrix.
Scoring matrices are used to determine the
relative score made by matching two
characters in a sequence alignment. These are
usually log-odds of the likelihood of two
characters being derived from a common
ancestral character. There are many flavors of
scoring matrices for amino acid sequences,
nucleotide sequences, and codon sequences,
and each is derived from the alignment of
"known" homologous sequences. These
alignments are then used to determine the
likelihood of one character being at the same
position in the sequence as another character.
38.
Scoring matrix fornucleotide sequence is simple. A positive value or high score
is given for match and a negative value or low score for mismatch.
Scoring matrix for amino acid are more complicated because scoring has not
only given to same amino acid residues but also given to those amino acids that
have same biochemical properties.
39.
K-TUPLE
More efficient thandynamic programming
E.g. BLAST and FASTA
BLAST : discovery of a unknown gene in the mouse, a scientist will typically
perform a BLAST with the human genome to see if human carry similar gene.
BLAST will identify sequences in the human genome that resemble to the mouse
gene based on similarity of sequence.
FASTA: text based format for representing nucleotide or protein sequence.