Scale Up & Post approval Changes and Bulk active chemical & Post approval changes
1.
SUBMITTED TO:
Dr. ManishKumar
Professor (HOD)
Department of Pharmaceutics
SUBMITTED BY:
Chirag Thakur
M Pharm 1st
Semester
(Pharmaceutical quality assurance)
SUBJECT NAME: PRODUCT DEVELOPMENT & TECHNOLOGY
TRANSFER(MQA104T)
SEMINAR TITLE: SUPAC & BACPAC
2.
INTRODUCTION
• Overview ofSUPAC (Scale-Up and Post-Approval Changes)
• Overview of BACPAC (Bulk Active Chemicals Post-Approval
Changes)
• Importance in regulatory compliance
• Difference between SUPAC and BACPAC
• First introduced in 1995 for immediate-release oral solid dosage forms
(SUPAC)
• Draft guidance published in 1999(BACPAC)
3.
REGULATORY BACKGROUND
• Issuedby the US FDA
• Ensures product quality post changes
• Covers manufacturing, equipment, site, and scale changes
• After approval of a drug product or drug substance, manufacturers may need
to make changes (scale-up, site change, new equipment, new supplier, etc.).
• Such changes could impact safety, efficacy, or quality, so regulatory agencies
provide guidelines.
• USFDA (CDER) developed SUPAC & BACPAC to streamline post-approval
changes.
4.
INTRODUCTION TO SUPAC
•SUPAC stands for Scale-Up and Post-Approval Changes.
• It is a regulatory guideline issued by the USFDA (Center for Drug
Evaluation and Research – CDER) in collaboration with AAPS.
• Introduced in 1995 for immediate-release oral solid dosage forms and
later expanded to modified-release, semisolid, and sterile products.
• The main purpose of SUPAC is to provide a clear regulatory pathway
for changes that occur after drug product approval.
• FDA issued various guidelines.
• SUPAC – IR , SUPAC – MR , SUPAC - SS.
5.
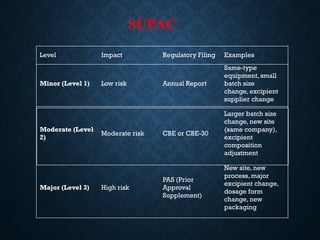
SUPAC
Level Impact RegulatoryFiling Examples
Minor (Level 1) Low risk Annual Report
Same-type
equipment, small
batch size
change, excipient
supplier change
Moderate (Level
2)
Moderate risk CBE or CBE-30
Larger batch size
change, new site
(same company),
excipient
composition
adjustment
Major (Level 3) High risk
PAS (Prior
Approval
Supplement)
New site, new
process, major
excipient change,
dosage form
change, new
packaging
6.
CHANGES IN SUPAC
Level1 – Minor Change
• A change that has minimal or no effect on the product’s quality, identity, strength,
purity, or potency.
• Regulatory Filing: Reported only in the Annual Report.
• Risk Level: Low risk.
• Examples:
• Change in equipment with the same design and principle (e.g., tablet press of the same
compression system).
• Batch size increase/decrease ≤10× the pilot/ biobatch size.
• Change in supplier of an excipient (if compendial and of the same grade).
7.
• Level 2– Moderate Change
• Definition:
A change that may have a moderate potential to impact product
quality/performance.
• Regulatory Filing:
• CBE (Changes Being Effected) Supplement → A change can be made
immediately after submission.
• CBE-30 → can be made 30 days after FDA submission (if no objection).
Examples: batch size increase/decrease beyond 10 times the
pilot/biobatch size (within the same equipment class).
• Change in manufacturing site .
• Change in equipment design but still same operating principle.
• Excipient quantitative changes .
8.
Level 3 changes:
A change that has a substantial potential impact on the product’s quality, safety,
or efficacy.
• Regulatory Filing: Requires Prior Approval Supplement (PAS) → must be
approved by FDA before implementation.
• Risk Level: High risk.
• Examples: New manufacturing site (outside firm or contractor).
• New manufacturing process (e.g., wet granulation → direct compression).
• Change in dosage form (tablet → capsule).
• Change in sterilization method (moist heat → irradiation).
• Change in container–closure system (glass → plastic, blister → bottle).
• New drug strength introduction.
9.
BACPAC
A change thathas a substantial potential impact on the product’s quality,
safety, or efficacy.
• Regulatory Filing: Requires Prior Approval Supplement (PAS) → must
be approved by FDA before implementation.
• Examples:
• New manufacturing site (outside firm or contractor).
• New manufacturing process (e.g., wet granulation → direct compression).
• Major change in formulation (addition/removal of excipients, large
excipient change).
• Change in dosage form (tablet → capsule).
• Change in sterilization method (moist heat → irradiation).
• Change in container–closure system (glass → plastic, blister → bottle).
10.
NEED OF BACPAC
BACPACis needed to regulate API manufacturing changes after
approval, ensuring quality, safety, and efficacy of the drug substance,
while giving manufacturers regulatory flexibility without requiring a
new drug application each time.
• Complexity of API Manufacturing
• Post-Approval Changes are Common
• Ensuring API Quality and Patient Safety
• Regulatory Flexibility in Change Control
• Harmonization with SUPAC
11.
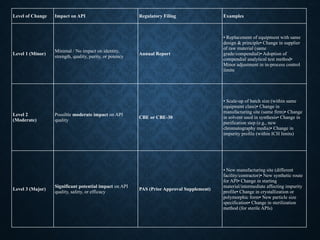
Level of ChangeImpact on API Regulatory Filing Examples
Level 1 (Minor)
Minimal / No impact on identity,
strength, quality, purity, or potency
Annual Report
• Replacement of equipment with same
design & principle• Change in supplier
of raw material (same
grade/compendial)• Adoption of
compendial analytical test method•
Minor adjustment in in-process control
limits
Level 2
(Moderate)
Possible moderate impact on API
quality
CBE or CBE-30
• Scale-up of batch size (within same
equipment class)• Change in
manufacturing site (same firm)• Change
in solvent used in synthesis• Change in
purification step (e.g., new
chromatography media)• Change in
impurity profile (within ICH limits)
Level 3 (Major)
Significant potential impact on API
quality, safety, or efficacy
PAS (Prior Approval Supplement)
• New manufacturing site (different
facility/contractor)• New synthetic route
for API• Change in starting
material/intermediate affecting impurity
profile• Change in crystallization or
polymorphic form• New particle size
specification• Change in sterilization
method (for sterile APIs)
12.
Moderate Changes (Level2)
• These changes may have a moderate potential to affect API quality and
therefore require FDA notification.
• Reported through CBE (Changes Being Effected) or CBE-30 submission:
• CBE: Can implement immediately after FDA submission.
• CBE-30: Must wait 30 days after submission unless FDA objects.
• Involve modifications in process, scale, or site, but without drastic changes in
critical properties.
• Might affect the impurity profile or stability, but within acceptable
ICH/compendial limits.
• Examples:
• Scale-up or scale-down of batch size (beyond 10× pilot/biobatch size) within the
same equipment class.
• Change in manufacturing site within same company or owned facilities.
• Change in solvent used in synthesis (if similar toxicity & compatibility).
• Modification in purification process (e.g., new chromatography resin/media).
13.
MINOR CHANGES (LEVEL1)
• These are low-risk changes that are unlikely to affect the identity, strength, quality, purity, or
potency of the API.
• They are reported only in the Annual Report, meaning FDA approval is not needed before
implementation.
• Usually involves substitutions with the same design/principle (no impact on process).
• Do not significantly alter the impurity profile, critical quality attributes, or specifications.
• Examples:
• Replacement of equipment with the same type and design (e.g., replacing a fluid bed dryer with
another of the same principle).
• Change in supplier of raw material (if compendial and same grade).
• Adoption of a compendial analytical method (e.g., USP method instead of in-house).
• Minor adjustments in in-process control limits (within validated ranges).
• Change in non-critical packaging material (not in direct contact with drug).
14.
Level 3 (Major)Changes – BACPAC
• High-risk changes → may significantly impact safety, efficacy, or quality.
• Require Prior Approval Supplement (PAS) → FDA approval before
implementation.
• Examples:
• New manufacturing site (different facility/contractor).
• New synthetic route for API.
• Change in starting material/intermediate affecting impurity profile.
• Change in crystallization process / new polymorphic form.
• New particle size specification or sterilization method.
15.
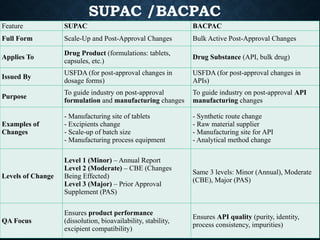
SUPAC /BACPAC
Feature SUPACBACPAC
Full Form Scale-Up and Post-Approval Changes Bulk Active Post-Approval Changes
Applies To
Drug Product (formulations: tablets,
capsules, etc.)
Drug Substance (API, bulk drug)
Issued By
USFDA (for post-approval changes in
dosage forms)
USFDA (for post-approval changes in
APIs)
Purpose
To guide industry on post-approval
formulation and manufacturing changes
To guide industry on post-approval API
manufacturing changes
Examples of
Changes
- Manufacturing site of tablets
- Excipients change
- Scale-up of batch size
- Manufacturing process equipment
- Synthetic route change
- Raw material supplier
- Manufacturing site for API
- Analytical method change
Levels of Change
Level 1 (Minor) – Annual Report
Level 2 (Moderate) – CBE (Changes
Being Effected)
Level 3 (Major) – Prior Approval
Supplement (PAS)
Same 3 levels: Minor (Annual), Moderate
(CBE), Major (PAS)
QA Focus
Ensures product performance
(dissolution, bioavailability, stability,
excipient compatibility)
Ensures API quality (purity, identity,
process consistency, impurities)
16.
CONCLUSION
• SUPAC andBACPAC guidelines ensure product consistency and safety
post-approval.
• Essential for regulatory compliance and patient safety.
• Ultimately, SUPAC and BACPAC support innovation, continuous
manufacturing, and global harmonization in the pharmaceutical
industry.
• BACPAC offers a flexible yet controlled framework, reducing
unnecessary submissions while focusing on significant changes.
17.
References
• FDA Guidancefor Industry: SUPAC-IR / SUPAC-MR / SUPAC-
SS(Available on U.S. FDA website – www.fda.gov
• Scale-up and Postapproval Changes (SUPAC) Regulations by P.
Sharma in Pharmaceutical Manufacturing Handbook: Regulations
and Quality (John Wiley) — discussion of SUPAC regulatory
framework in a handbook format.
• Pharmaceutical Quality Assurance book, Dr. Manish Kumar ,
Rajneesh Garg