Download to read offline

















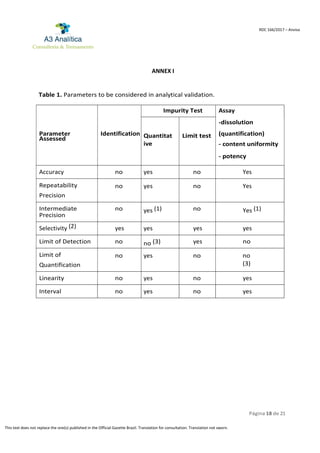




This document provides guidelines for validating analytical methods used in various stages of pharmaceutical production. It defines key terms related to analytical validation and outlines parameters that should be considered, including selectivity, limit of detection, precision, accuracy, and range. Methods must demonstrate they can reliably identify and quantify analytes without interference from other substances. Both validated and partial validation studies are described. Reference materials and characterization reports are also addressed.



































































