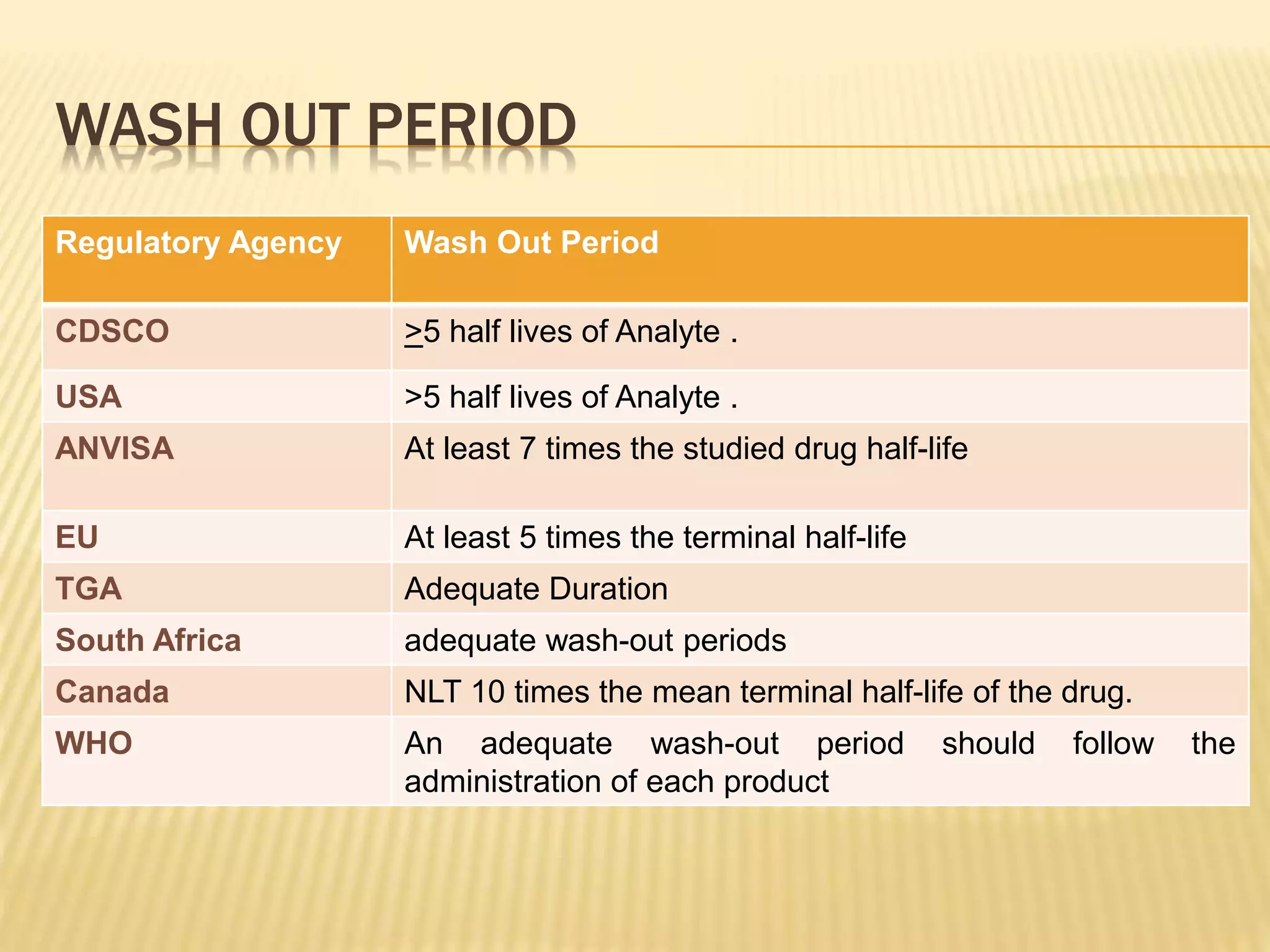
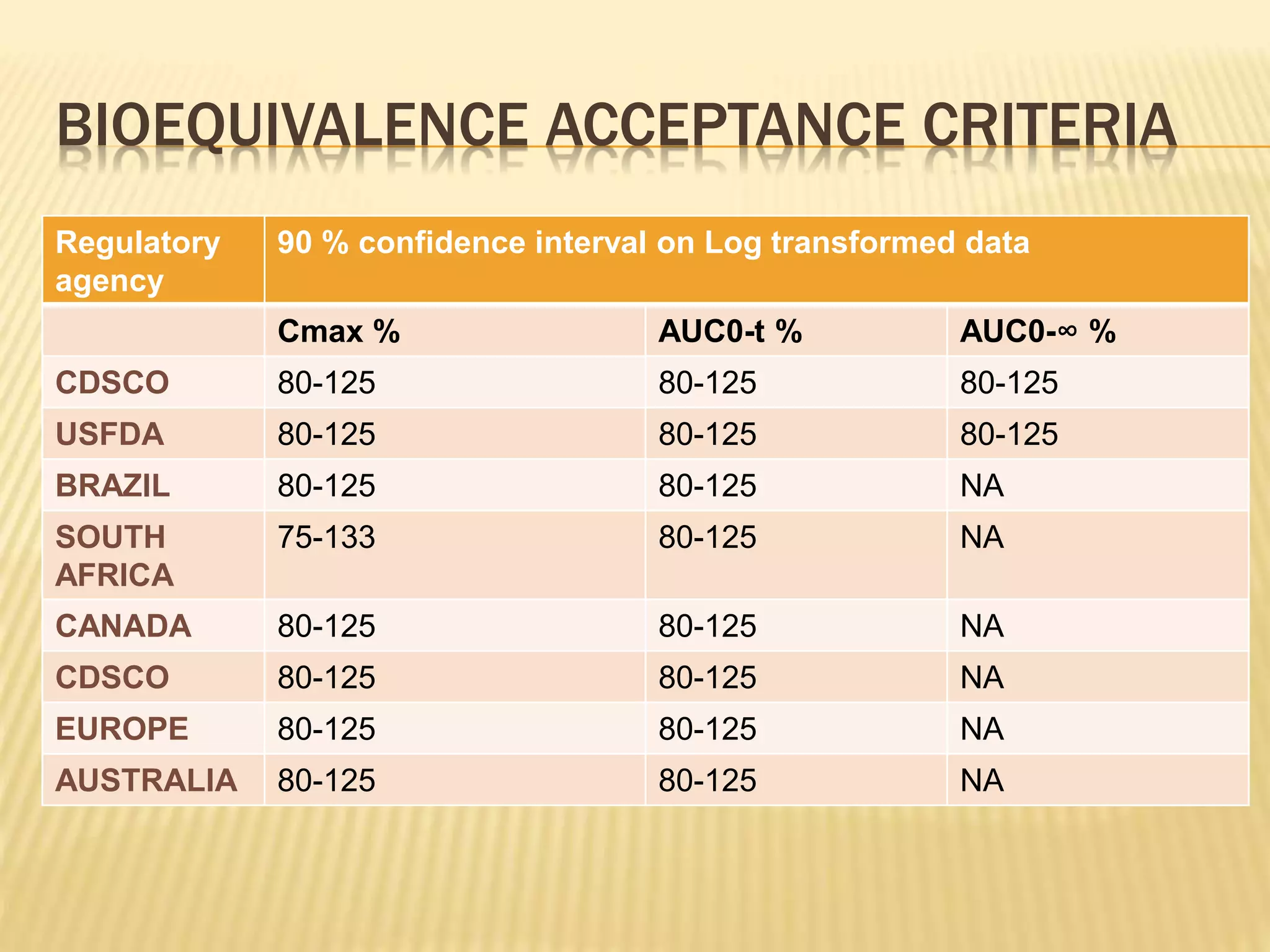
The document discusses guidelines for clinical trial protocols according to various regulatory agencies. It provides definitions of a protocol, describes common components of protocols such as objectives, study design, and safety assessments. It also outlines regulatory requirements for bioequivalence studies from agencies like FDA, EMA, and CDSCO regarding issues like study design, sample size, acceptance criteria, product handling and more. Requirements for conducting fed and fasting studies are also covered.













































![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)





























