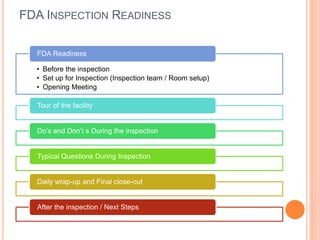
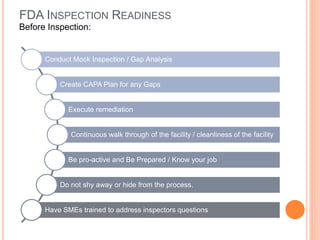
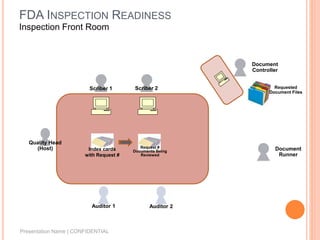
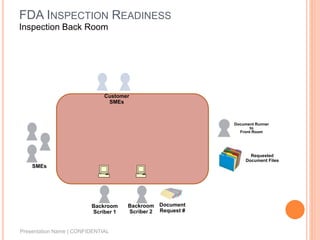
The document provides guidance on preparing for and managing an FDA inspection. It outlines steps to take before, during, and after the inspection including conducting a mock inspection, setting up an inspection team, and documenting any deficiencies found. It also provides dos and don'ts for interacting with inspectors such as being prepared, transparent, and avoiding arguments.





































































![Handling of a fda inspection [compatibility mode]](https://cdn.slidesharecdn.com/ss_thumbnails/handlingofafdainspectioncompatibilitymode-150610120404-lva1-app6891-thumbnail.jpg?width=640&height=640&fit=bounds)





































