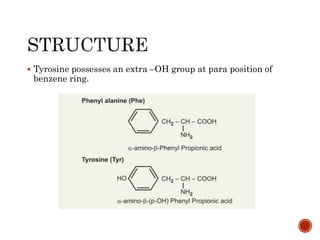
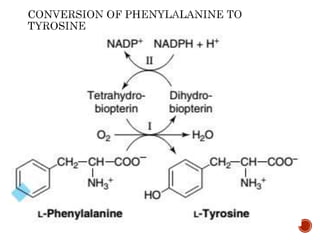
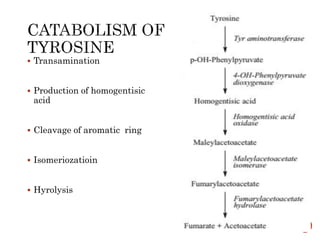
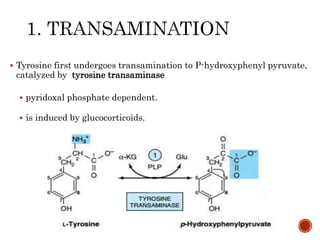
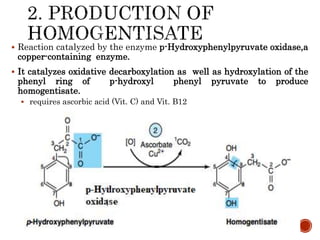
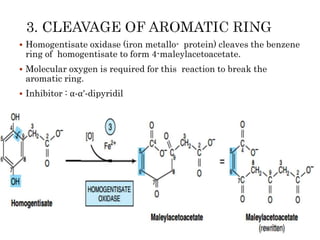
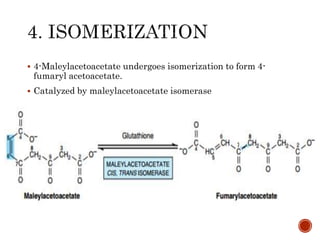
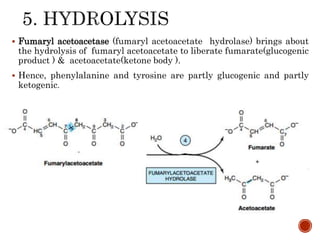
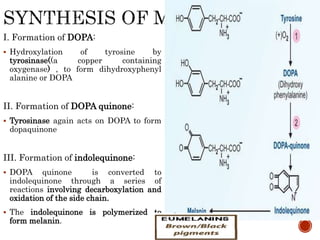
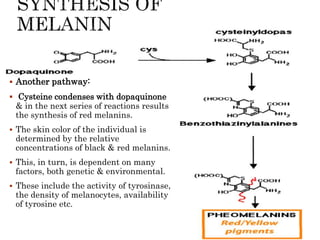
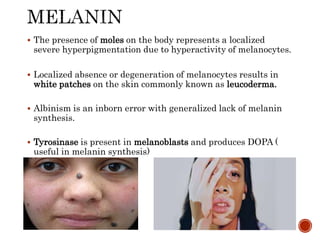
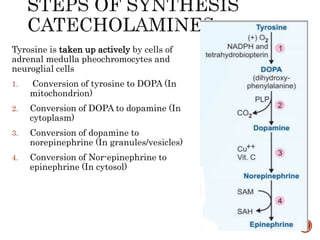
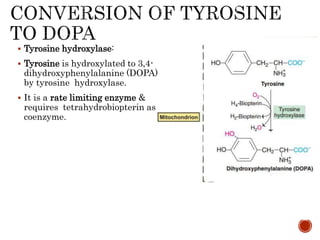
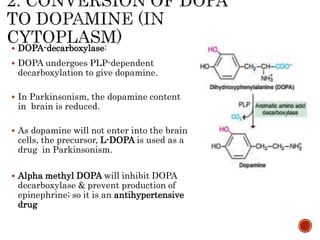
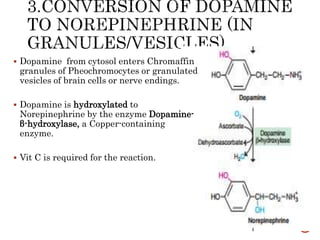
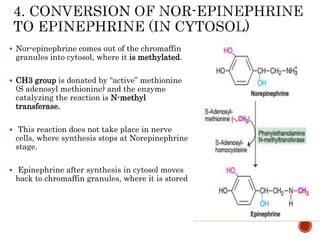
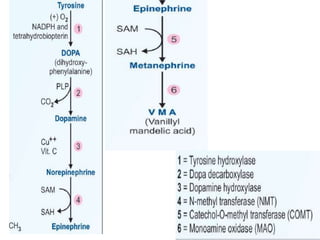
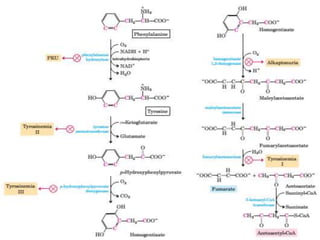
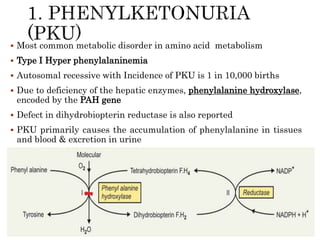
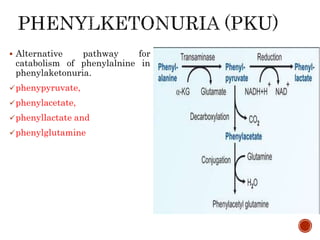
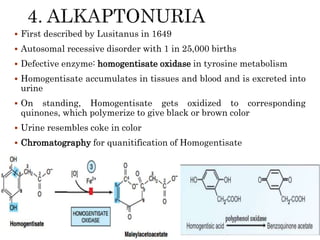
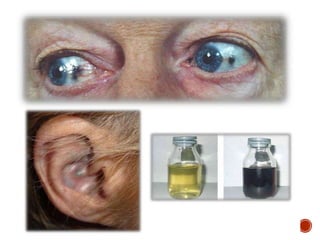
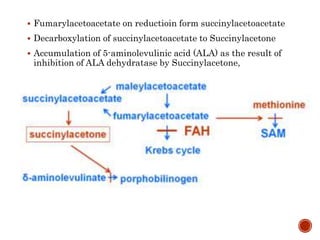
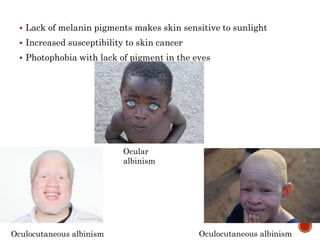
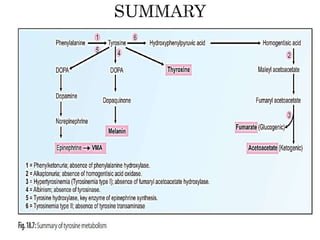
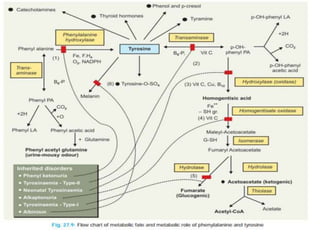
This document summarizes the structures, metabolism, and roles of phenylalanine and tyrosine. It discusses how phenylalanine is converted to tyrosine in the liver by the enzyme phenylalanine hydroxylase. Tyrosine can then be used to synthesize important compounds like thyroid hormones, melanin, and catecholamines. The document also outlines several disorders related to phenylalanine and tyrosine metabolism, including phenylketonuria, tyrosinemia, alkaptonuria, and albinism.
















![1.Effect on CNS
Mental retardation, failure to walk or talk, failure of
growth, seizures and tremor
Hypotyrosinemia:low[tyrosine],low neurotransmitters
(loss of biogenic amines at critical stages in postnatal
brain maturation)
Defective brain myelination (chronic and irreversible)](https://image.slidesharecdn.com/phenylalanineandtyrosineforclass-191108022520/85/Phenylalanine-and-tyrosine-for-class-36-320.jpg)




















































![Amino_acid_metabolisnm_3rd_unit[1] by BKG sir.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/aminoacidmetabolisnm3rdunit1-230618103839-68f88ba5-thumbnail.jpg?width=640&height=640&fit=bounds)
![Amino_acid_metabolisnm_3rd_unit[1] by bulet sir.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/aminoacidmetabolisnm3rdunit1-230620073918-9d18680e-thumbnail.jpg?width=640&height=640&fit=bounds)






















![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)






![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)









