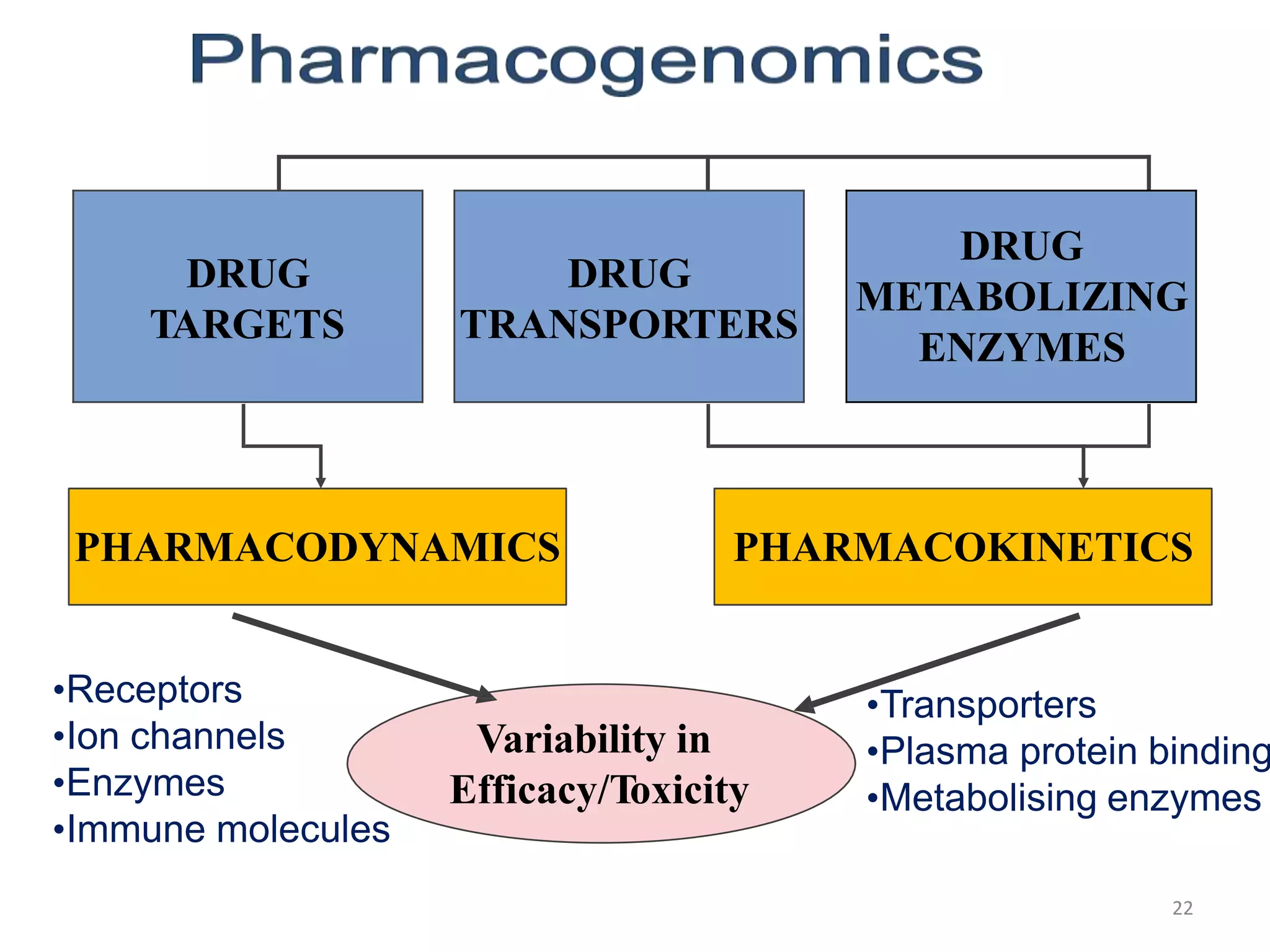
This document discusses the history and principles of pharmacogenetics. It begins by defining pharmacogenetics as the study of inherited genetic differences in drug metabolism pathways that affect individual drug responses. It then discusses key terms like pharmacogenomics and personalized medicine. The document outlines several genetic variations that can impact drug metabolism, such as variations in cytochrome P450 enzymes and N-acetyltransferase. It also discusses how these genetic variations can result in differences in drug efficacy and toxicity between individuals.













![ Human genome has 30,000 genes.
Each gene has several thousands of nucleotides.
Each person inherits 2 copies of genes one from each
parent.
Any two individuals DNA is 99.9% identical
3 billion nucleotides.
Variation is seen in >1% of population called
polymorphism
Of that most common is SNP
BASICS IN GENETICS
Between 2 people (except identical twins) the rate of
genetic variation (individuality) is about 0.1%
[0.1% of 3 billion = 3 million base pair differences]
14](https://image.slidesharecdn.com/pharmacogeneticsbydr-181002035250/75/Pharmacogenetics-by-dr-mahi-14-2048.jpg)