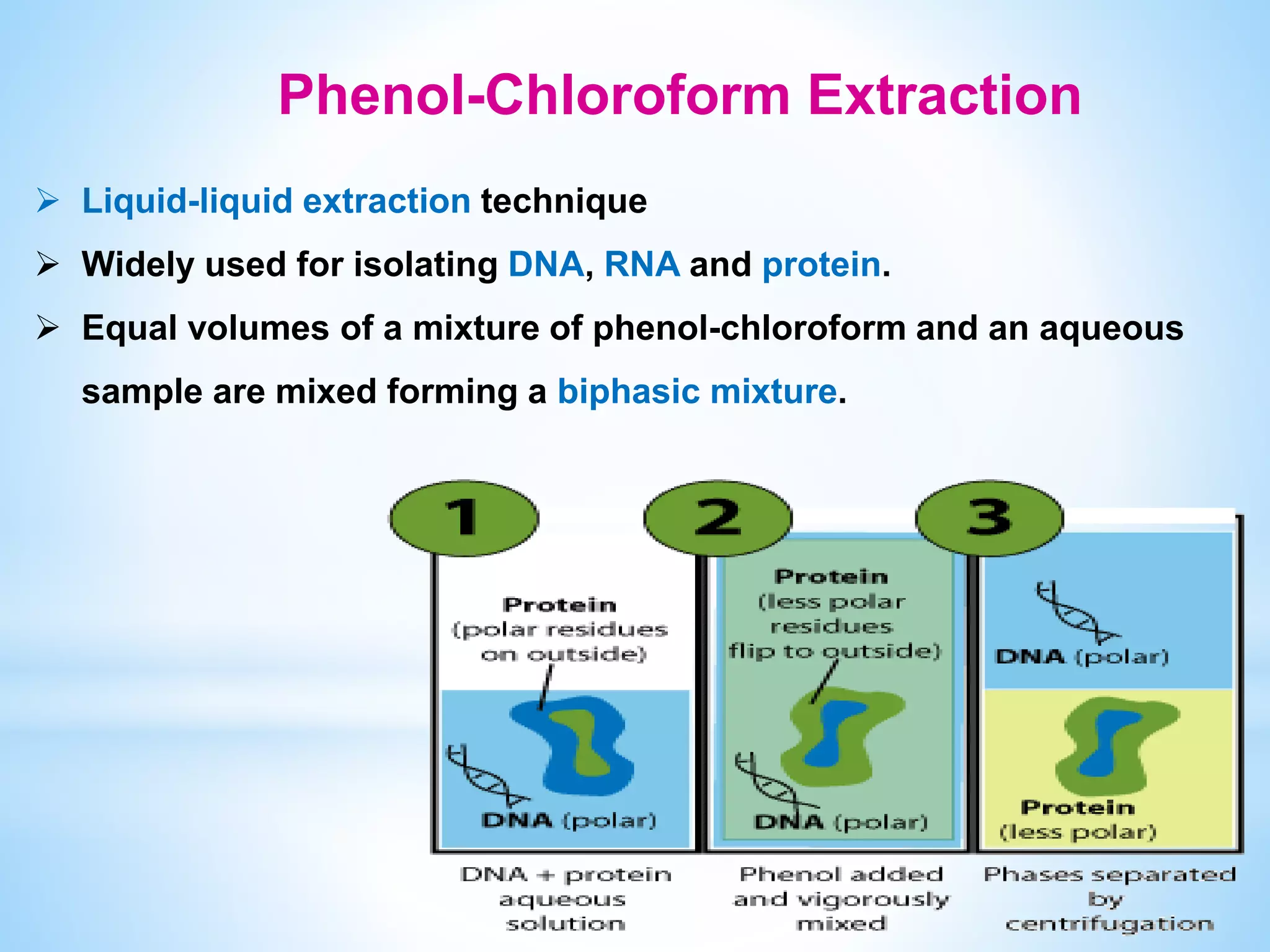
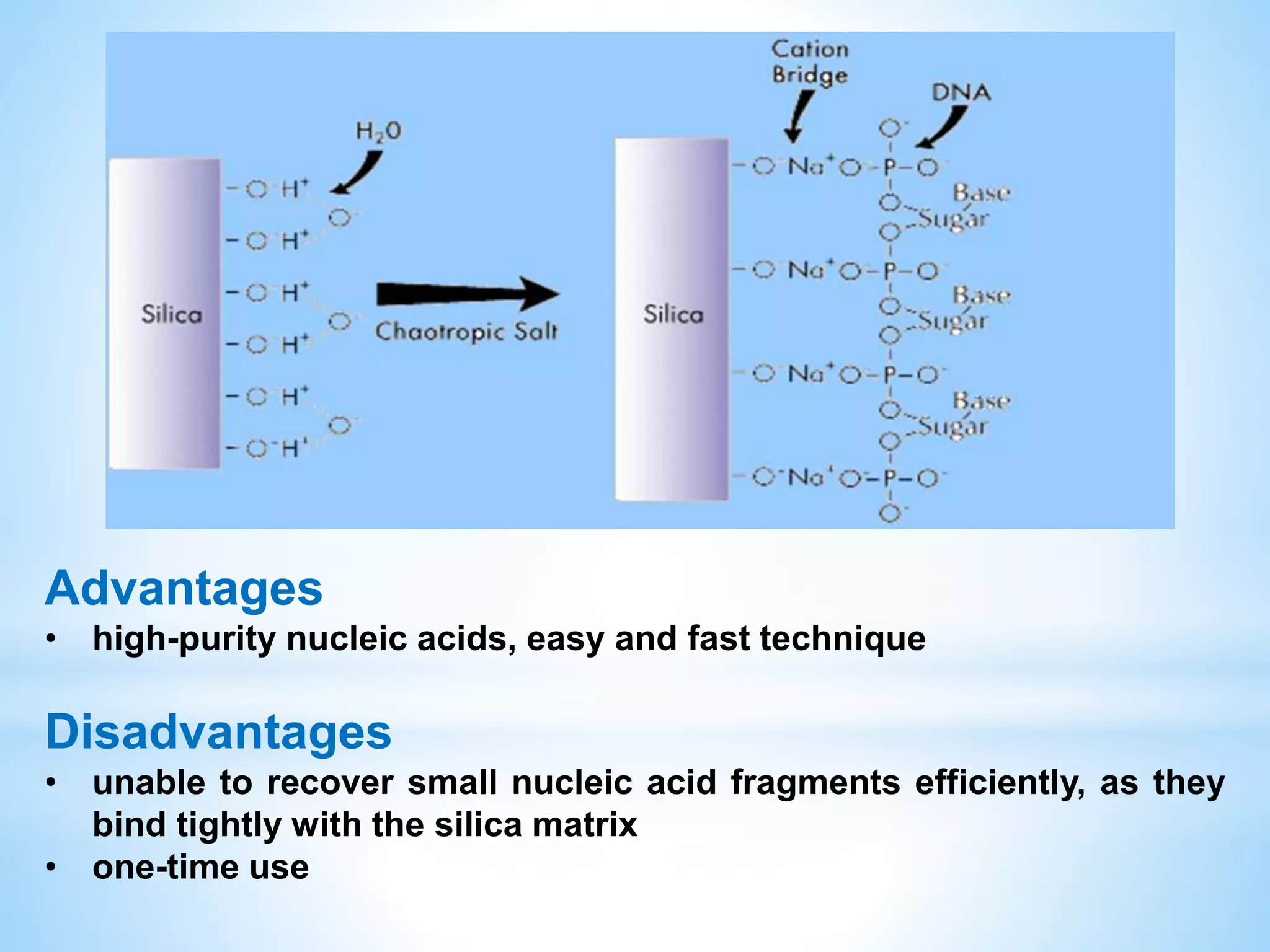
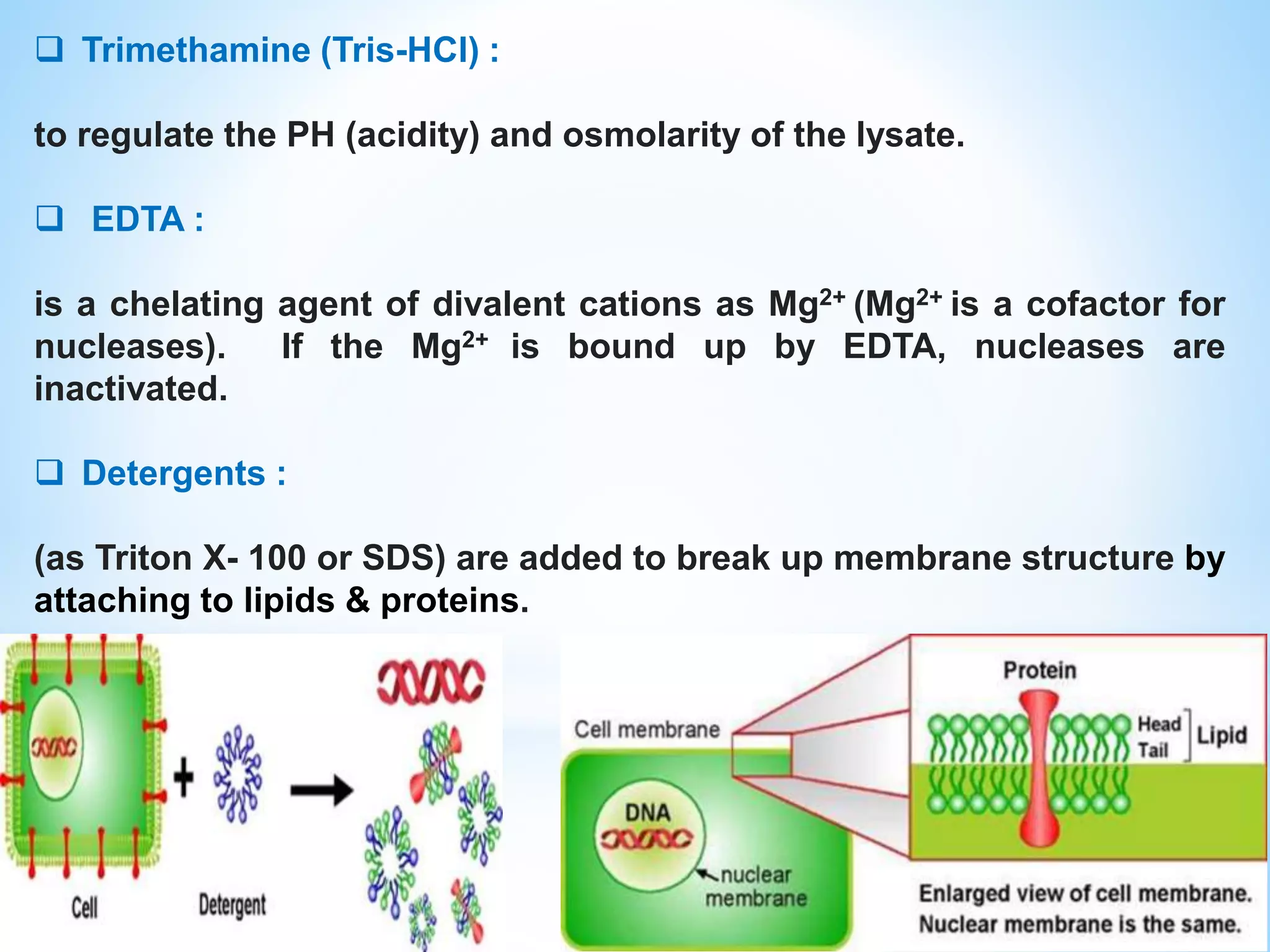
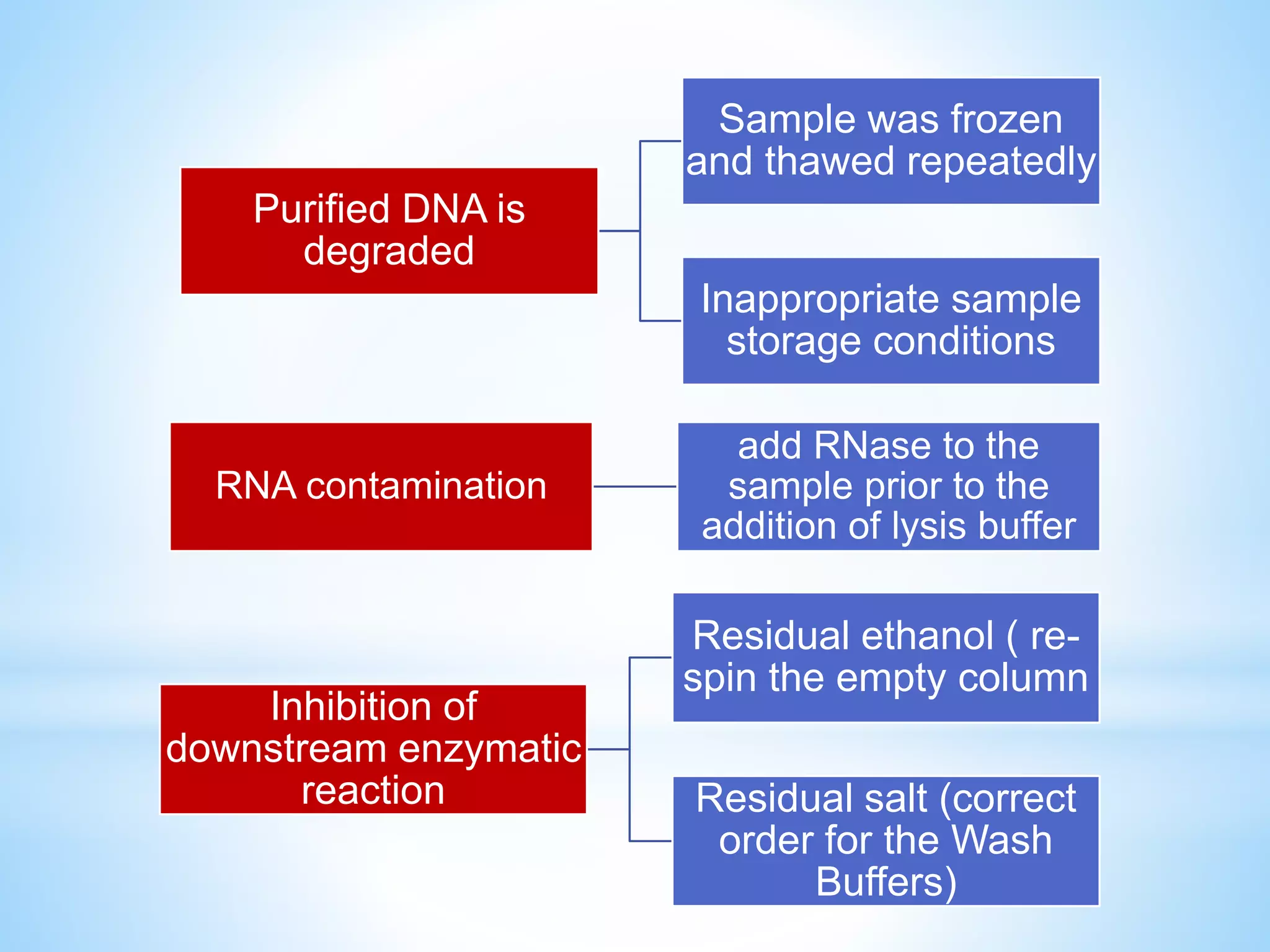
The document describes various methods for extracting nucleic acids, with a focus on DNA extraction. It discusses the basic extraction process, which involves lysing cells, breaking down proteins and RNA, and separating nucleic acids from cellular debris. Specific extraction techniques are covered, including non-organic extraction using proteinase K and salting out, organic extraction using phenol-chloroform, and spin column purification. Key steps and considerations for DNA extraction from blood samples are also outlined.