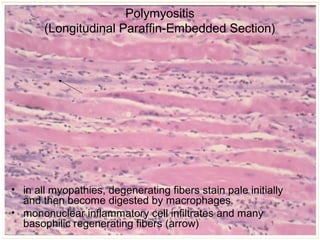
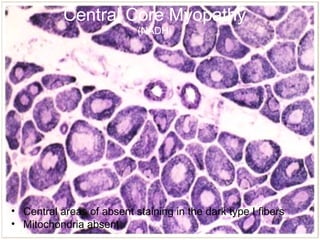
This document summarizes skeletal muscle disorders and the histopathological findings seen in muscle biopsy specimens. It describes the normal muscle histology and classifications of acquired and inherited myopathies. Key points include the inflammatory features of polymyositis and dermatomyositis, rimmed vacuoles and inclusions in inclusion body myositis, and the absence of dystrophin staining in Duchenne muscular dystrophy. Overall, the document provides an overview of muscle biopsy findings and their significance in diagnosing different skeletal muscle disorders.






















































![Emery-Dreifuss Muscular
Dystrophy
• Caused by mutation in genes that encode nuclear lamina proteins.
• Triad
– Slowly progressive humeroperoneal weakness
– Cardiomyopathy
– Early contractures of the Achilles tendon, spine & elbow
• X-linked form [EMD1]– mutation in genes encoding emerin
• Autosomal form [EMD2] – mutation in genes encoding lamin
• These protein helps in maintaining the shape and mechanical stability of the
nucleus during muscle contraction.](https://image.slidesharecdn.com/myopathy1-230402095148-48e9669b/85/myopathy-1-pdf-57-320.jpg)



























































































