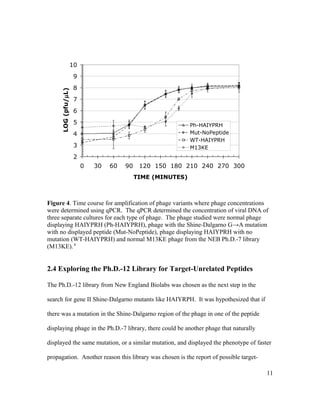
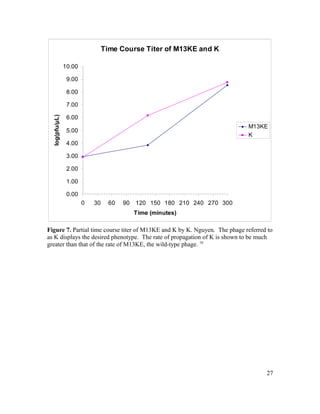
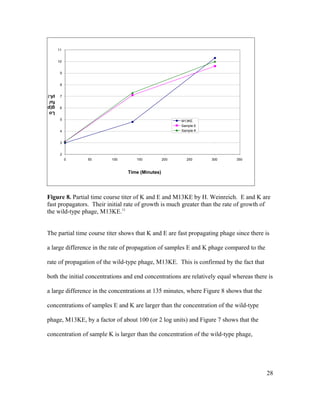
This document is Michelle Houle's senior thesis which explores using the Ph.D.-12 peptide phage display library to identify bacteriophage that propagate faster than average. The thesis provides background on bacteriophage and phage display libraries. Houle then serially amplified the Ph.D.-12 library for 3 rounds to enrich for fast propagating phage. 48 phage clones were isolated and sequenced. Two clones were identified as fast propagators based on time course titers and both had mutations near the Shine-Dalgarno region of gene II.