Download to read offline





![Parameter Finding
Cellularity
Markedly
hypercellular (75–
90%)
Myeloid:
Erythroid Ratio
Increased (10–
30:1)
Blasts <10% in CP
Megakaryocytes
Increased but
normal
morphology
Fibrosis
Typically absent
or minimal
Test Purpose
Karyotyping
Detect Philadelphia
chromosome (t[9;22]
[q34;q11])
FISH
(Fluorescence
In Situ
Hybridization)
Detect BCR-ABL
fusion gene
RT-PCR
(Quantitative
PCR)
Measure BCR-ABL1
transcript levels
using International
Scale (IS) for
monitoring response
to treatment
3. Bone Marrow Biopsy 4. Cytogenetics and Molecular Diagnostics](https://image.slidesharecdn.com/chronicleukemia-250713170042-4b8fbffe/85/Leukemia-CHRONIC-LEUKEMIA-CML-CLL-pptx-11-320.jpg)









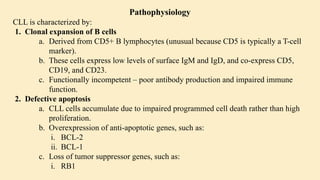


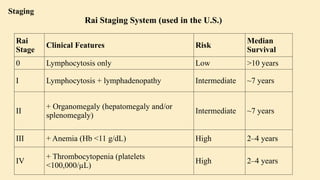








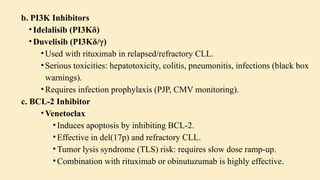



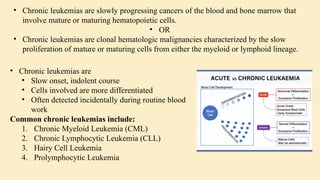
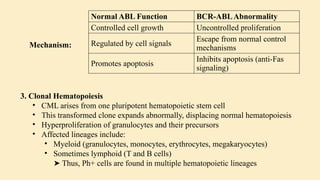
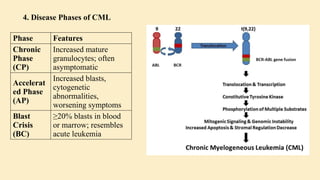
Chronic leukemias are slowly progressing cancers of the blood and bone marrow that involve mature or maturing hematopoietic cells. Unlike acute leukemia, which is aggressive and rapidly fatal without treatment, chronic leukemias progress over months or years and may be asymptomatic for a long time. 🔑 Key Features: Slow onset, indolent course Cells involved are more differentiated Often detected incidentally during routine blood work Includes: Chronic Myeloid Leukemia (CML) – Myeloid origin Chronic Lymphocytic Leukemia (CLL) – Lymphoid origin Hairy cell leukemia Prolymphocytic leukemia 🔷 2. What is Chronic Myeloid Leukemia (CML)? CML is a clonal stem cell disorder classified as a myeloproliferative neoplasm. It results in the uncontrolled proliferation of mature and maturing granulocytes (neutrophils, basophils, eosinophils) in the blood and bone marrow. 🔑 Key Characteristics: Involves a pluripotent hematopoietic stem cell Strongly associated with a genetic abnormality: the Philadelphia chromosome Leads to bone marrow hyperplasia and leukocytosis Progresses in phases: Chronic phase Accelerated phase Blast crisis 🔷 3. Etiology (Cause) of CML CML is mainly caused by a specific genetic mutation rather than environmental or inherited factors. ✅ Known Facts: Philadelphia chromosome (Ph) formation causes CML This chromosome is produced by a translocation between: Chromosome 9 (ABL gene) Chromosome 22 (BCR gene) ❌ No association with: Heredity or family history Geography Ethnicity or race Socioeconomic status ⚠️ Associated Risk Factor: Exposure to ionizing radiation Seen in atomic bomb survivors (Hiroshima and Nagasaki) Higher incidence in patients exposed to therapeutic radiation 🔷 4. Epidemiology of CML 📊 Statistics: ~8,990 new CML cases in the US (2019) Accounts for 15–20% of adult leukemias More common in men than women 👤 Patient Profile: Median age at diagnosis: 67 years Rare in children; more common in adults 🔷 5. Pathophysiology of CML 🔹 A. Philadelphia Chromosome (Ph) Discovered in 1960 Caused by reciprocal translocation: t(9;22)(q34;q11) Creates the BCR-ABL fusion gene 🔹 B. BCR-ABL Fusion Gene The fusion gene codes for a protein with tyrosine kinase activity The most common abnormal protein: p210 BCR-ABL Constitutively active (always “on”) → uncontrolled cell proliferation 🧬 Mechanism: Normal ABL Function BCR-ABL Abnormality Controlled cell growth Uncontrolled proliferation Regulated by cell signals Escape from normal control mechanisms Promotes apoptosis Inhibits apoptosis (anti-Fas signaling) 🔹 C. Effect on Bone Marrow and Blood Hyperproliferation of granulocytes and their precursors Overcrowding leads to suppression of normal hematopoiesis In later stages: cytopenias and marrow fibrosis 🔹 D. Clonality and Pluripotent Origin Disease arises from one mutated stem cell Affects myeloid and lymphoid progenitors (seen in early progenitor cells) Leads to monoclonal expansion of malignant























































































