Download to read offline




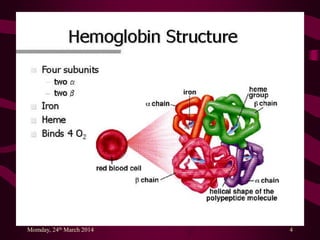



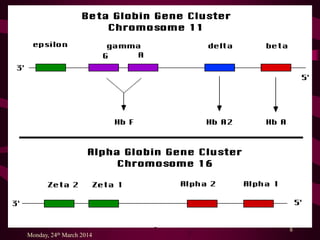













The document discusses thalassemias, hereditary hypochromic anemias caused by genetic defects affecting hemoglobin synthesis. It details alpha and beta thalassemias, their genetic causes, clinical presentations, and treatment options, including the importance of managing complications. The document also highlights emerging therapies such as bone marrow transplants, gene therapy, and dietary interventions.
































































![THALASEMIA definition and pathophysiologyd].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/thalasemia-240515044127-8ce804c5-thumbnail.jpg?width=640&height=640&fit=bounds)





















![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)


![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)















