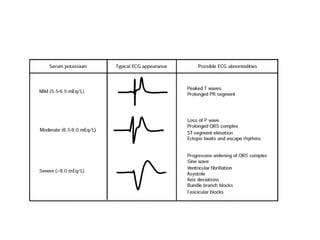
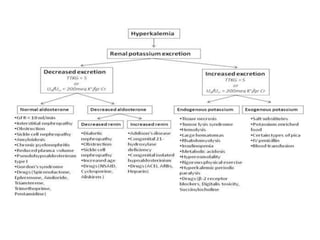
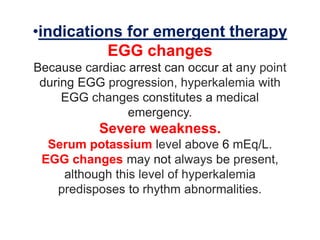
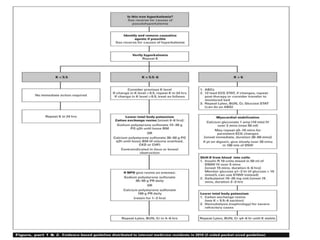
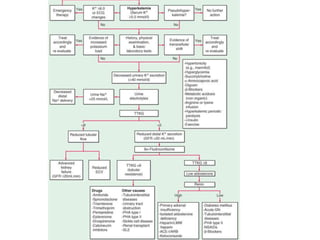
Hyperkalemia can be caused by increased potassium intake, redistribution of potassium from cells into the bloodstream, or decreased excretion of potassium by the kidneys. Clinical manifestations range from weakness to cardiac arrhythmias. Treatment involves membrane stabilization with calcium, shifting potassium into cells with insulin and glucose or sodium bicarbonate, and removing potassium from the body with loop diuretics, potassium-binding resins, or hemodialysis. Glucose should be given with insulin to treat hyperkalemia only if blood sugar is below 175 mg/dL.


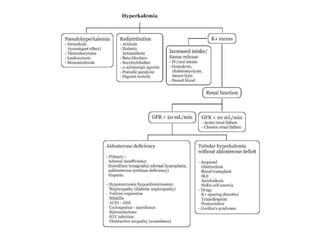





![States of hypoaldosteronism
include :
Decreased renin-angiotensin system
Activity
(e.g., hyporeninemic hypoaldosteronism
in diabetes, interstitial nephritis, ACE
inhibitors, nonsteroidal antiinflammatory
drugs [NSAIDs], cyclosporine)](https://image.slidesharecdn.com/neehyperkalamia-161018073954/85/hyperkalamia-11-320.jpg)