Downloaded 180 times





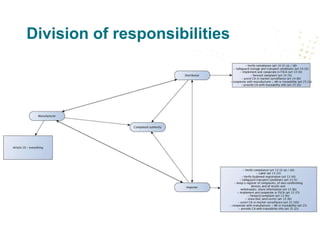


![New stuff in Blue Guide that is
often overlooked
• The role of fulfilment houses (FHs) as service provider to EOs (section
3.4)
• If their activities “go beyond those of parcel service providers that
provide clearance services, sorting, transport and delivery of
parcels […] they should be considered as distributors and should
fulfil the corresponding legal responsibilities.”
• Many medical devices companies find that their FHs are not ‘neutral’
service providers but regulated distributors ex art. 14 MDR / IVDR as a
result of the services they provide](https://image.slidesharecdn.com/medtechsummiteconomicoperators2018-180614104552/85/Economic-operators-under-the-MDR-and-IVDR-12-320.jpg)



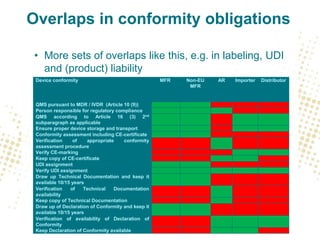












The document discusses the responsibilities and compliance obligations of economic operators in the medical device supply chain under the MDR and IVDR regulations. It outlines the distinctions between EEA and third countries, emphasizes the importance of post-market surveillance, and highlights the evolving role of authorized representatives and fulfillment houses. Additionally, it addresses potential impacts of Brexit on regulatory frameworks and the need for manufacturers to adapt their operations accordingly.






















![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)






















































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)









