

























![Clinical performance / efficacy?
• Medicinal products trials: “ascertaining its (their) safety and/or
efficacy”
• Medical devices [currently]: “demonstrate the safety and performance
of their devices”
• MDR proposal: inconsistent language
– Explanatory note 3.6: “performance of the clinical evaluation needed to
demonstrate the safety and performance of their devices”
– (34) „clinical investigation‟ means any systematic investigation in one or
more human subjects, undertaken to assess the safety or performance of
a device;
• But:
– Article 26 requires “summary of safety and clinical performance”](https://image.slidesharecdn.com/euhottopicsalliancepresentation-3-130605030552-phpapp02/85/Eu-hot-topics-alliance-presentation-3-27-320.jpg)


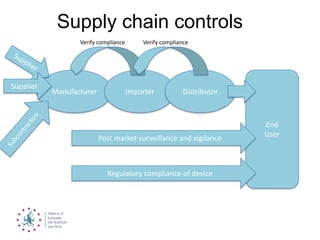











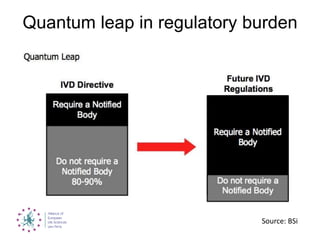






















This document summarizes recent EU regulatory proposals and developments regarding medical devices, in vitro diagnostics, and clinical trial data transparency. For medical devices, key proposals include stricter rules for notified bodies, more clinical data requirements, and alignment of some terminology with pharmaceuticals. For in vitro diagnostics, conformity assessment will depend more on risk class. Clinical trial data may become publicly available after marketing approval. Overall, the proposals aim to increase transparency but could significantly increase regulatory burden for these industries.






































































































