Downloaded 66 times






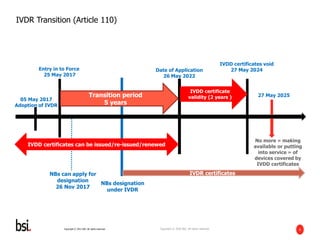
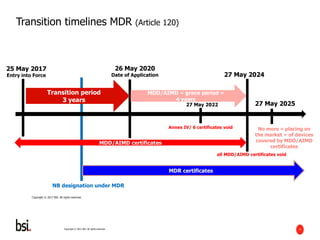
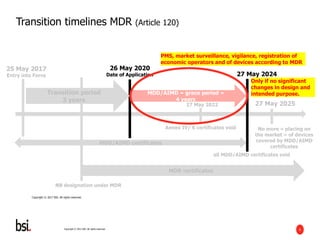
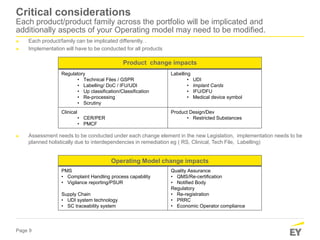
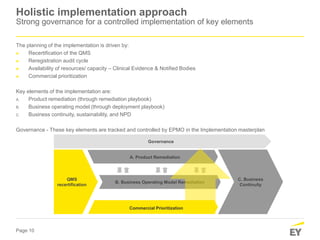

The document discusses the transition requirements and deadlines related to the EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR), emphasizing the need for conformity assessments by the end of the transitional periods. Key considerations include the lack of grandfathering provisions and the necessity for companies to reassess their devices for compliance under the new regulations. A holistic implementation approach is outlined, focusing on governance, product remediation, business operating model adjustments, and the importance of resource allocation.


































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)










