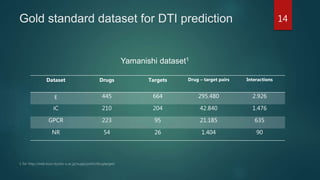
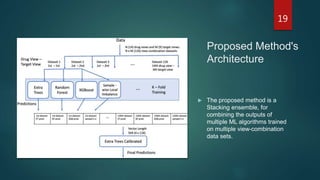
- The document proposes a multi-view stacking ensemble method for drug-target interaction (DTI) prediction that combines predictions from multiple machine learning models trained on different drug and target feature view combinations.
- It generates 126 view combination datasets from 14 drug views and 9 target views, then trains extra trees, random forest, and XGBoost classifiers on each view combination. Predictions from these base models are then combined using a stacking ensemble with an extra trees meta-learner.
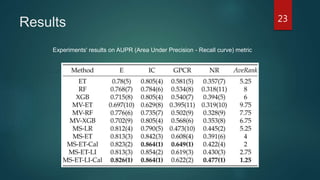
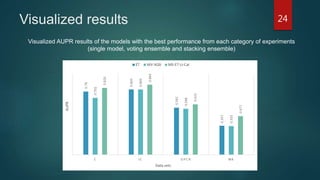
- The method is shown to outperform single models and voting ensembles, and calibration of the meta-learner and use of local imbalance measures provide further improvements to predictive performance on DTI prediction tasks.