Download as PDF, PPTX




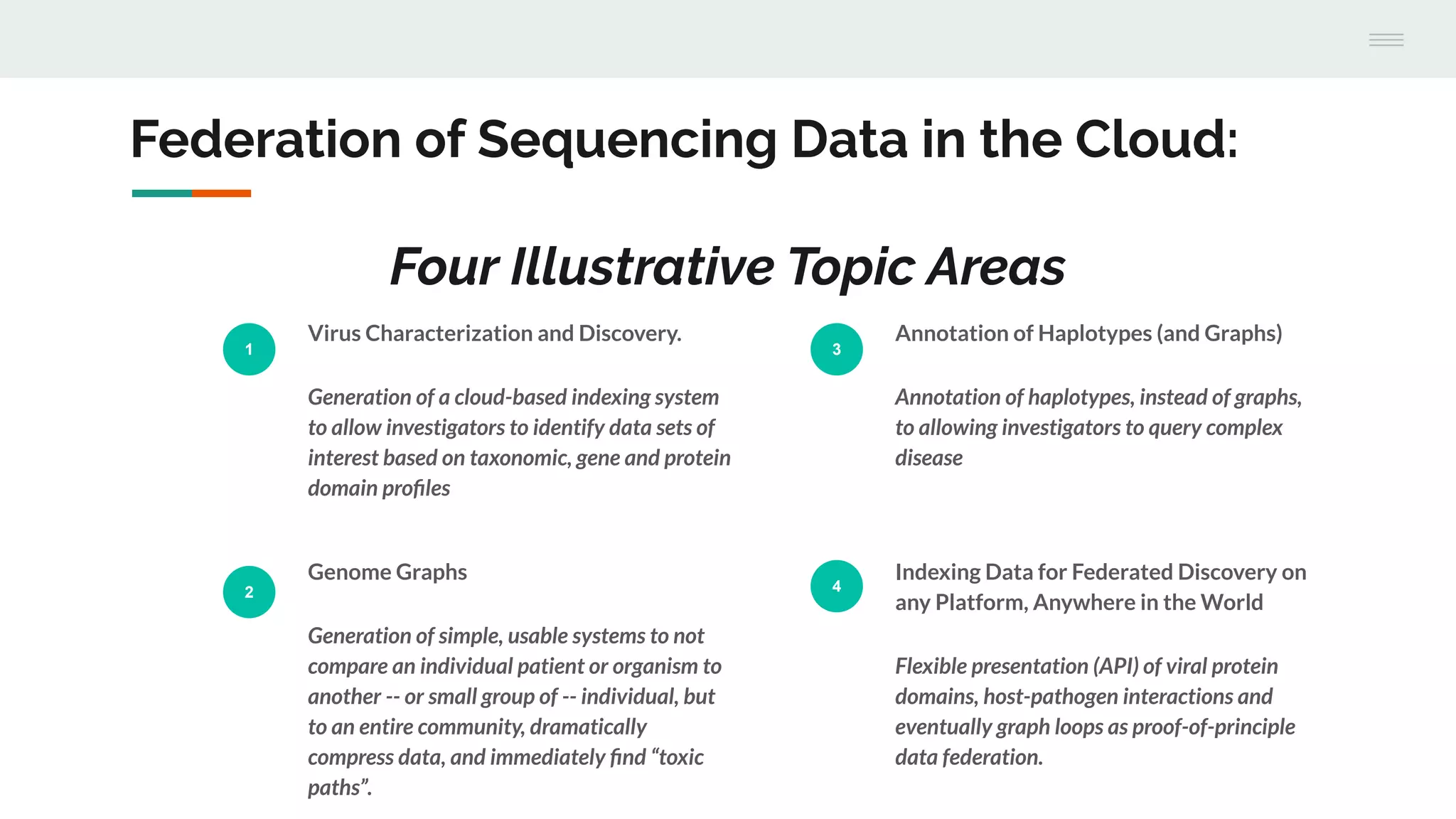
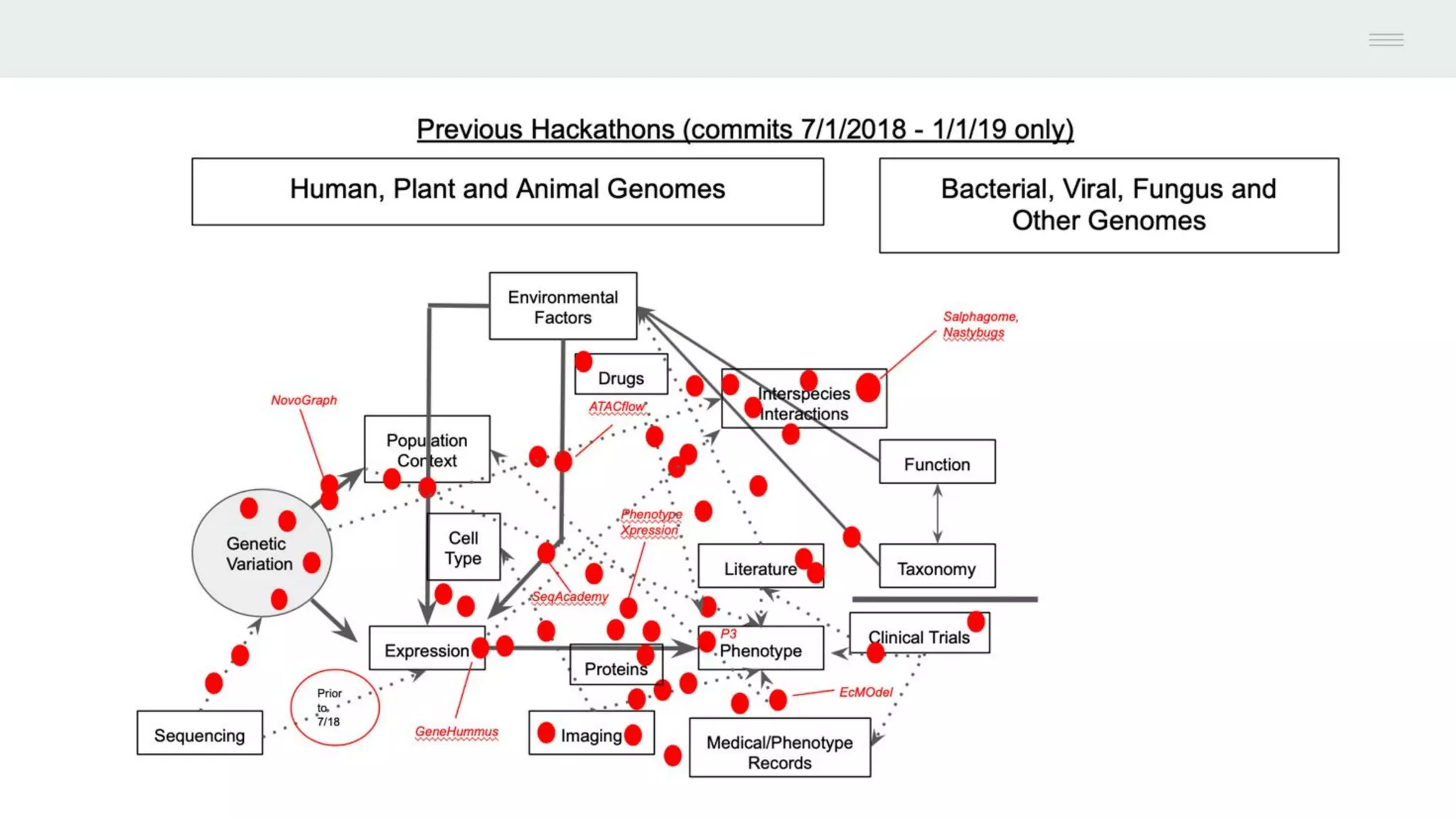
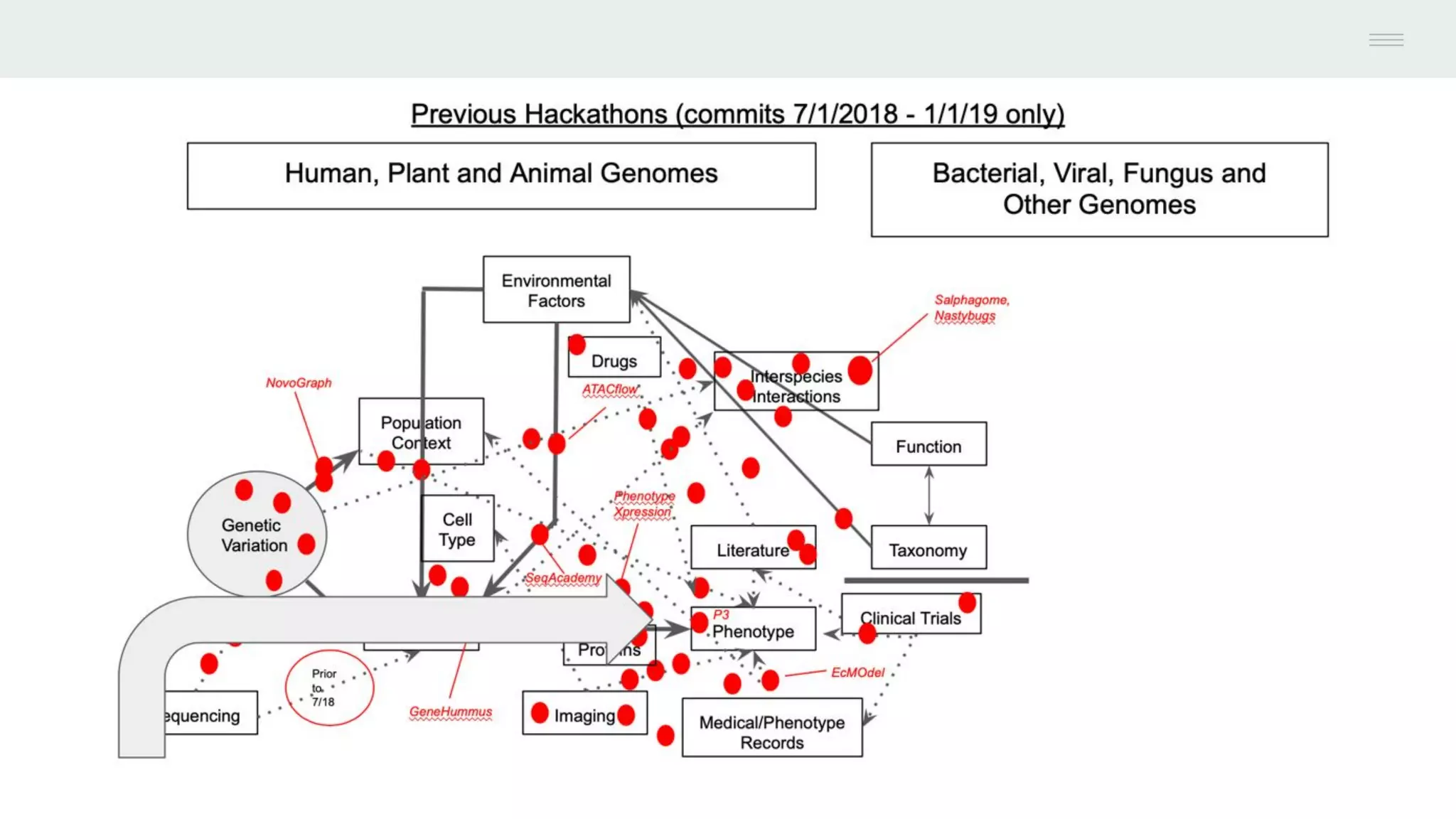
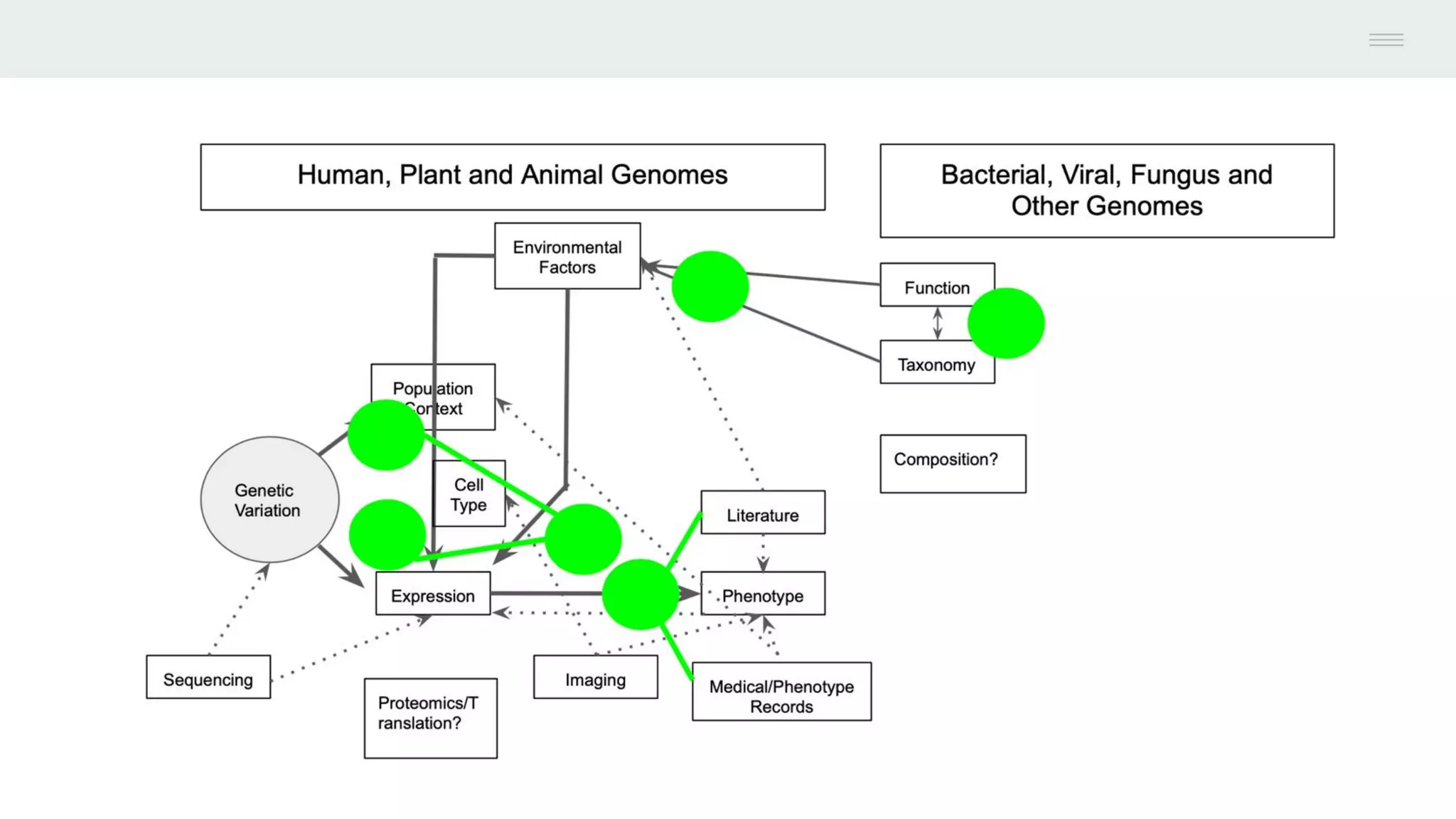

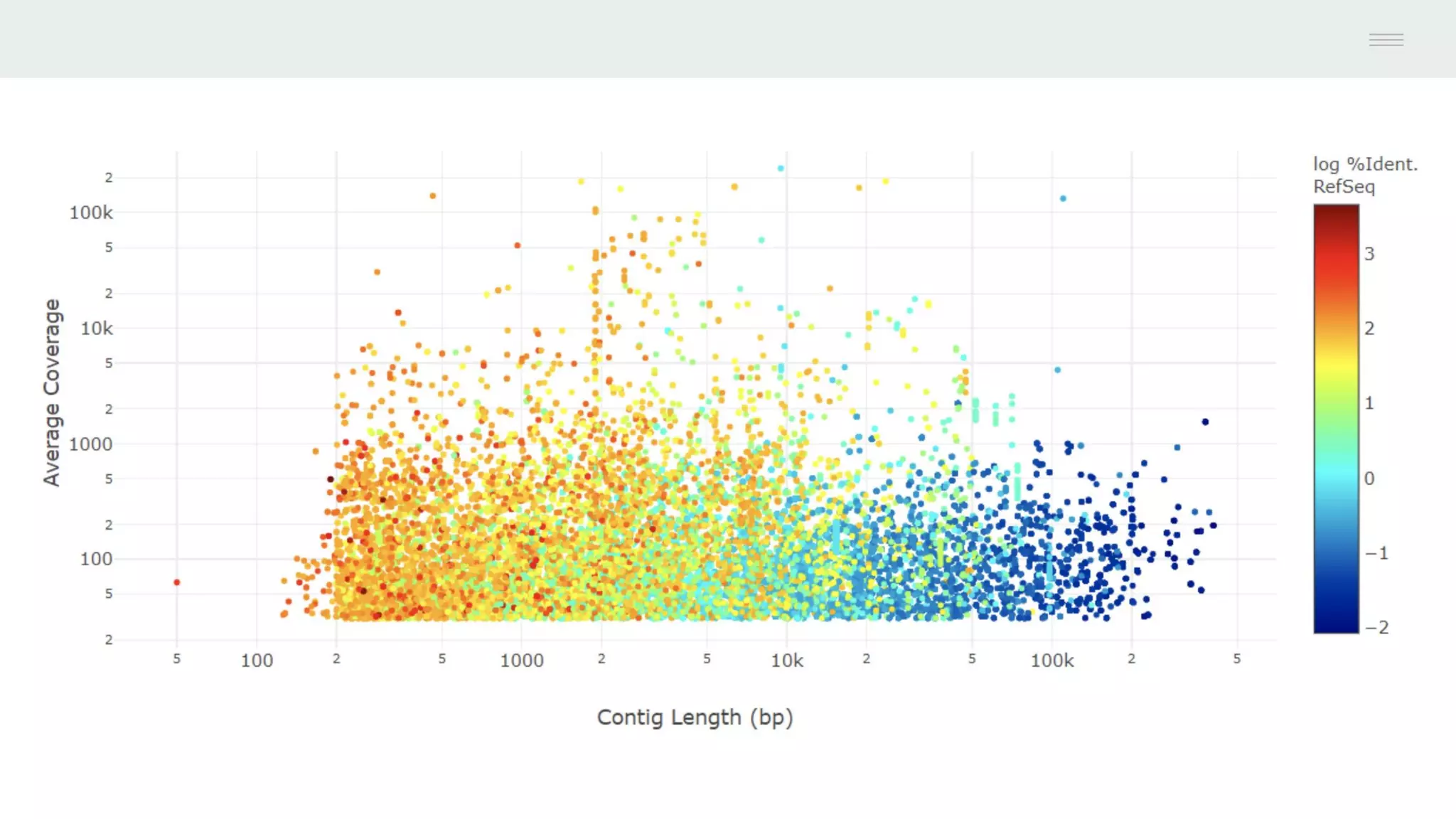
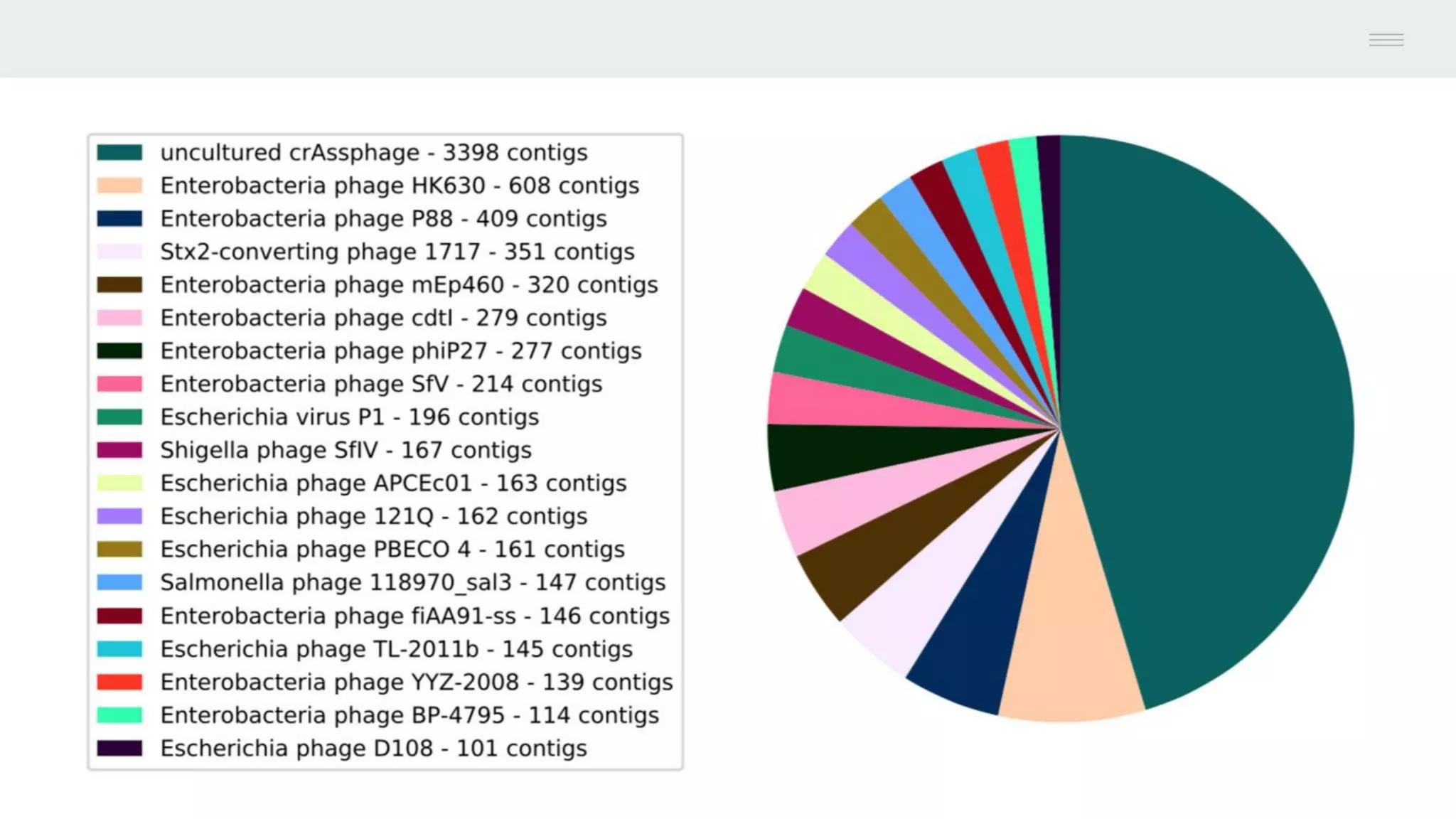


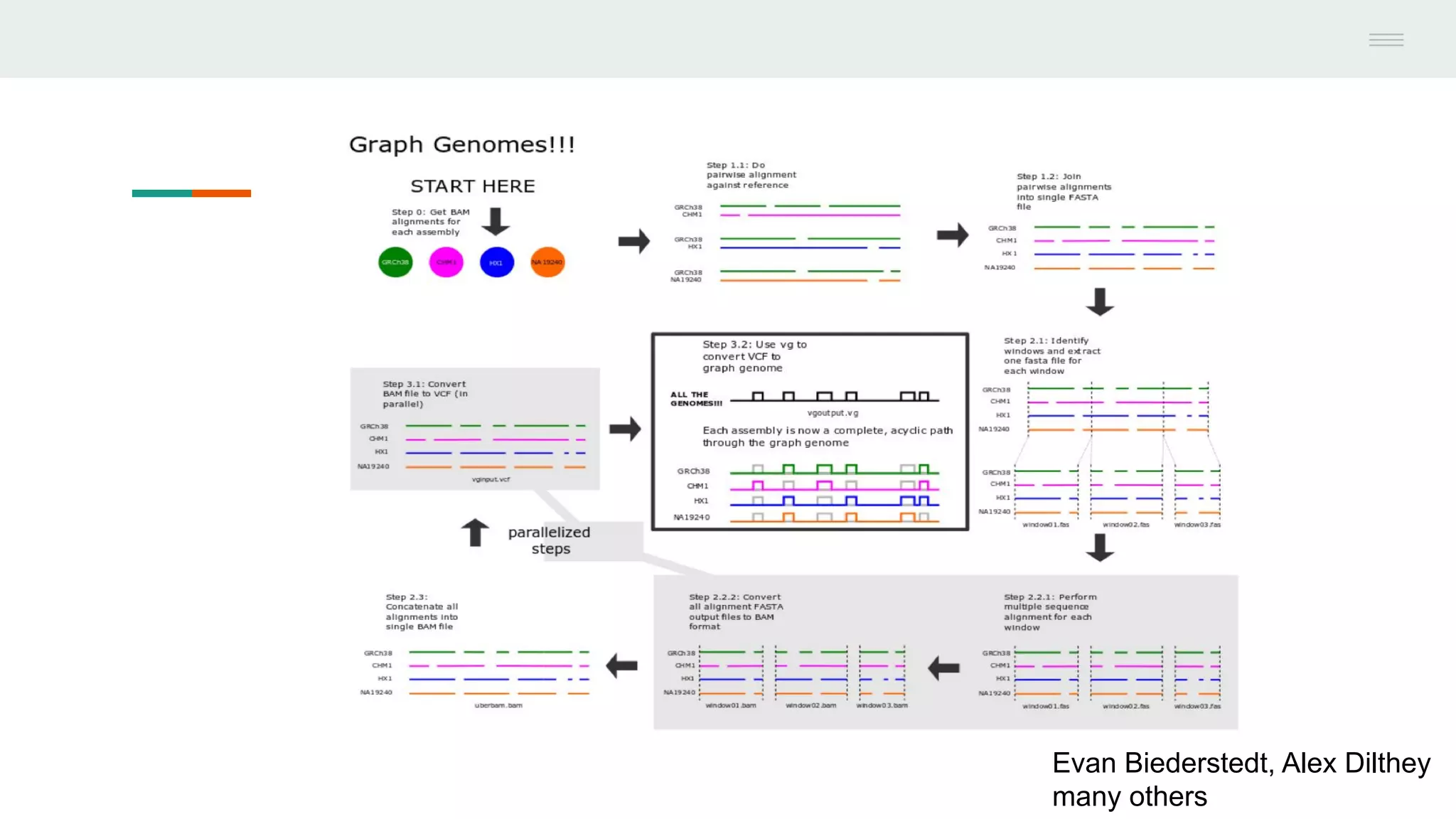
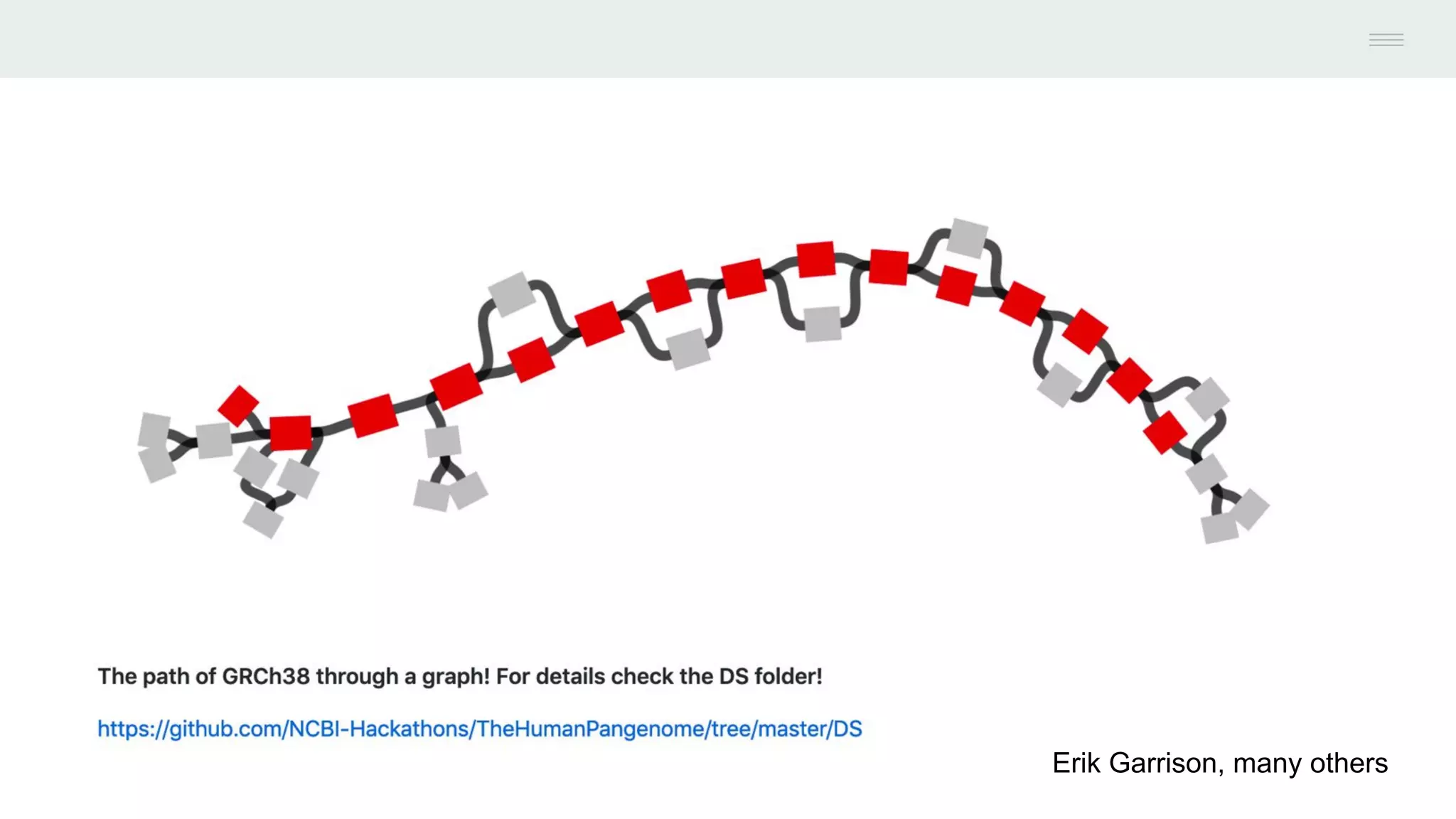
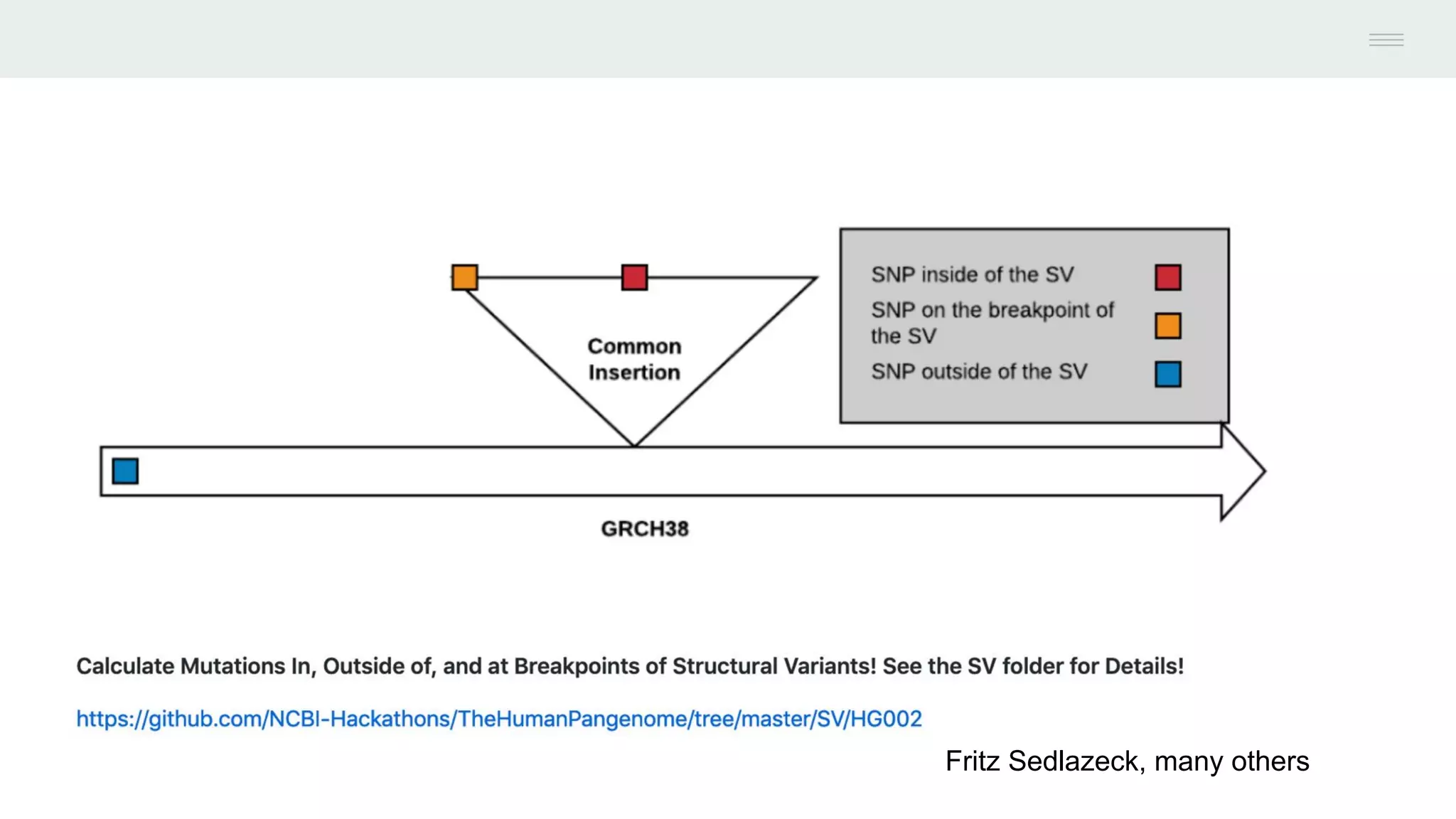

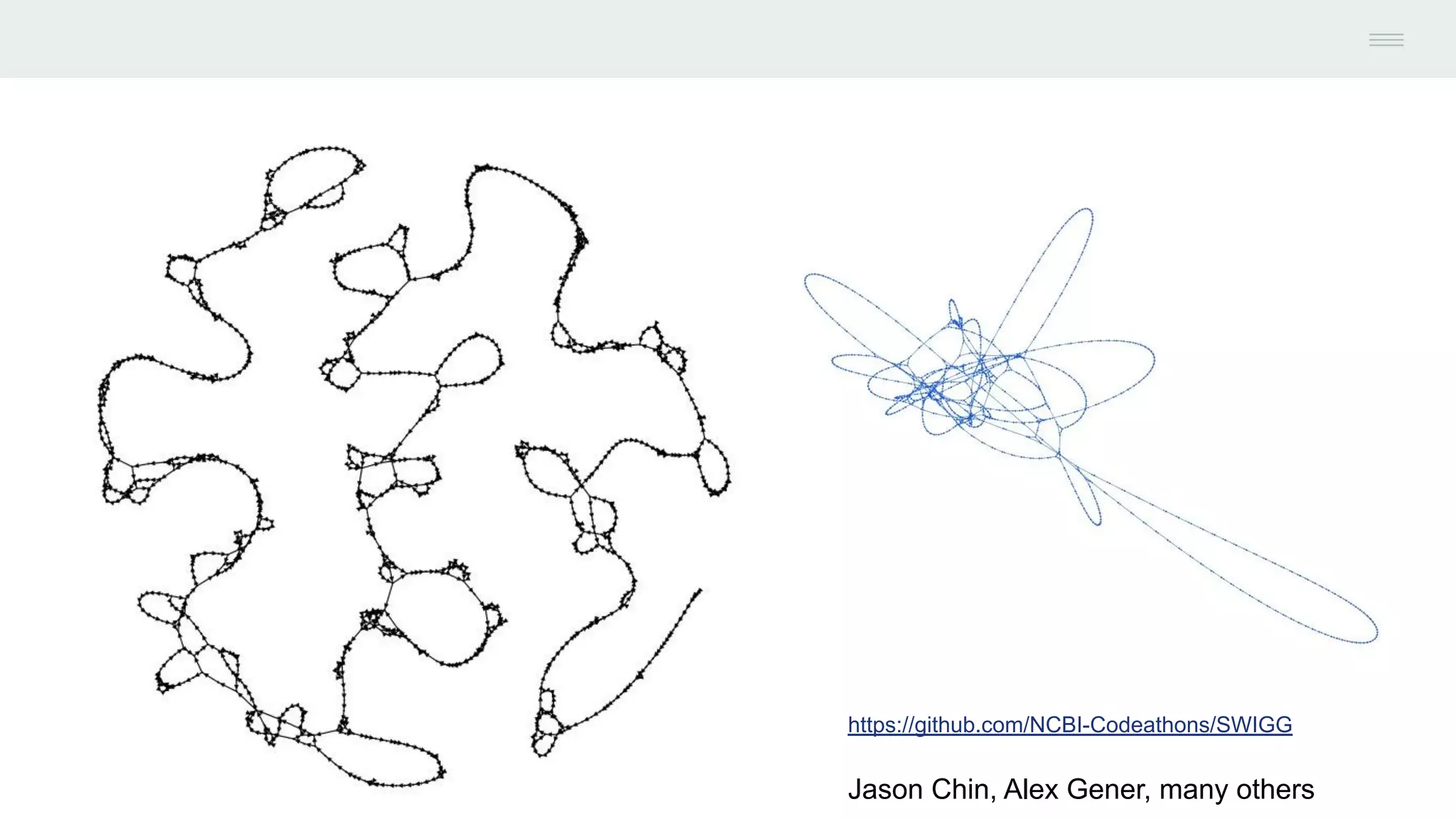

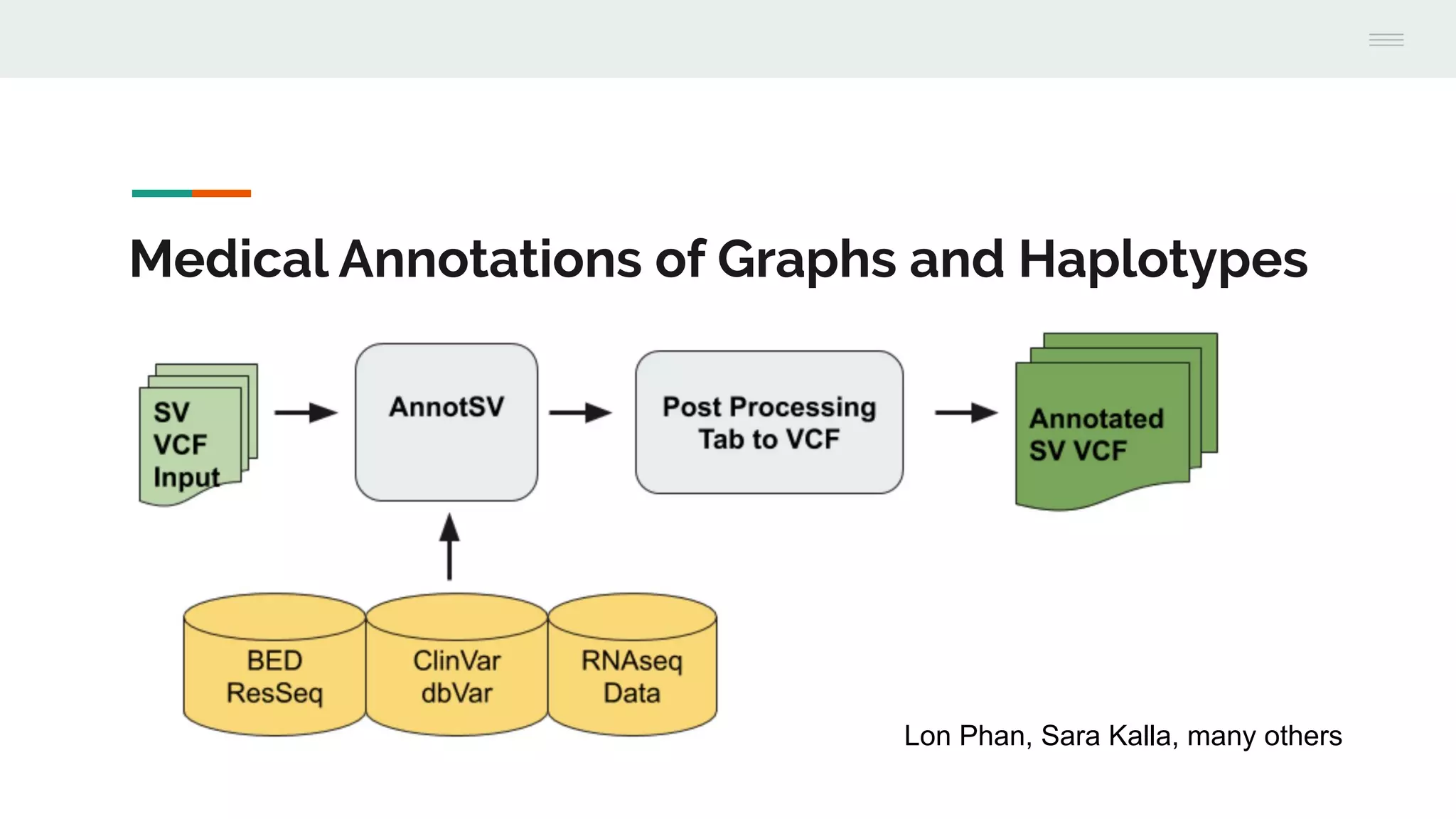
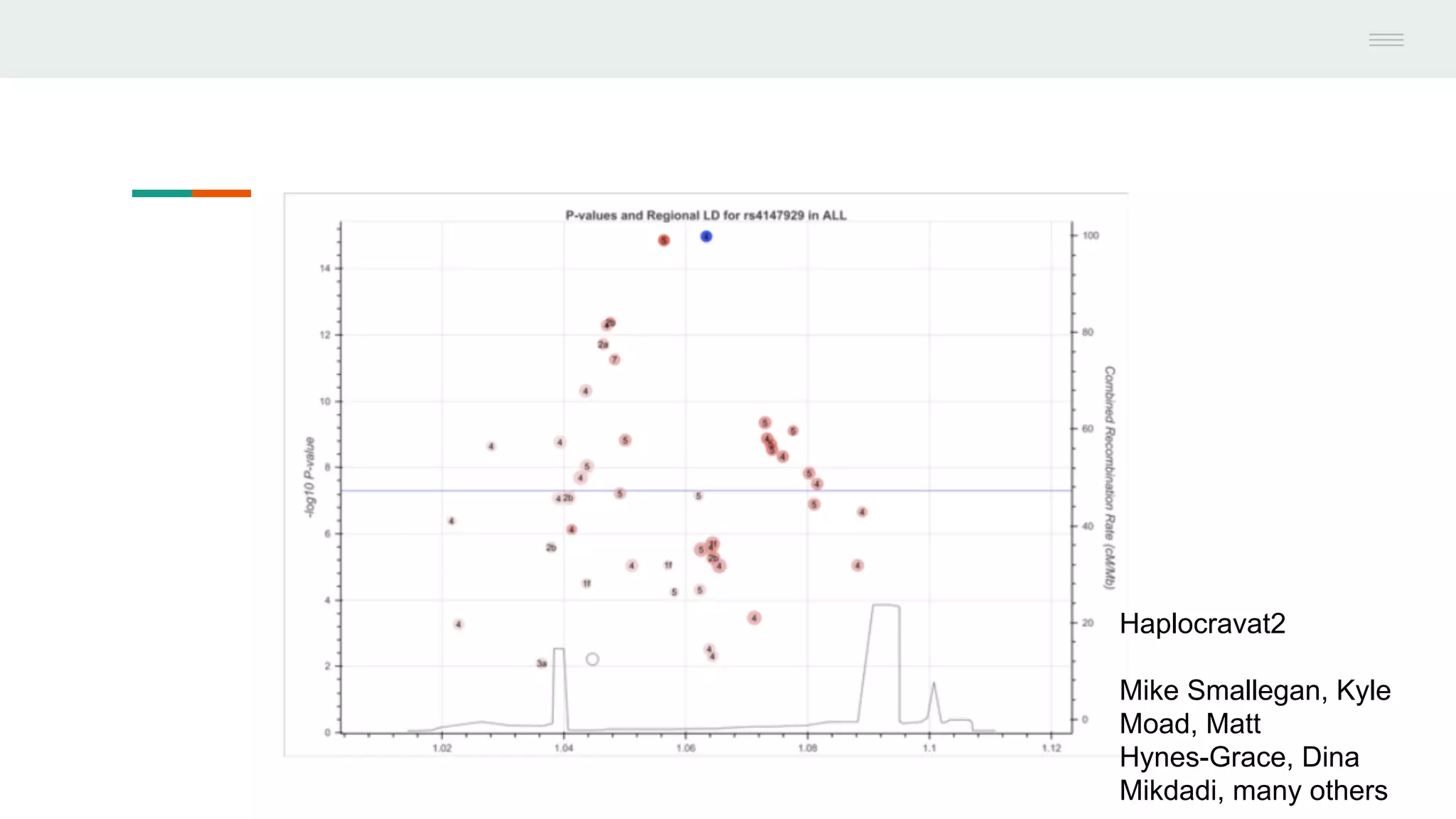
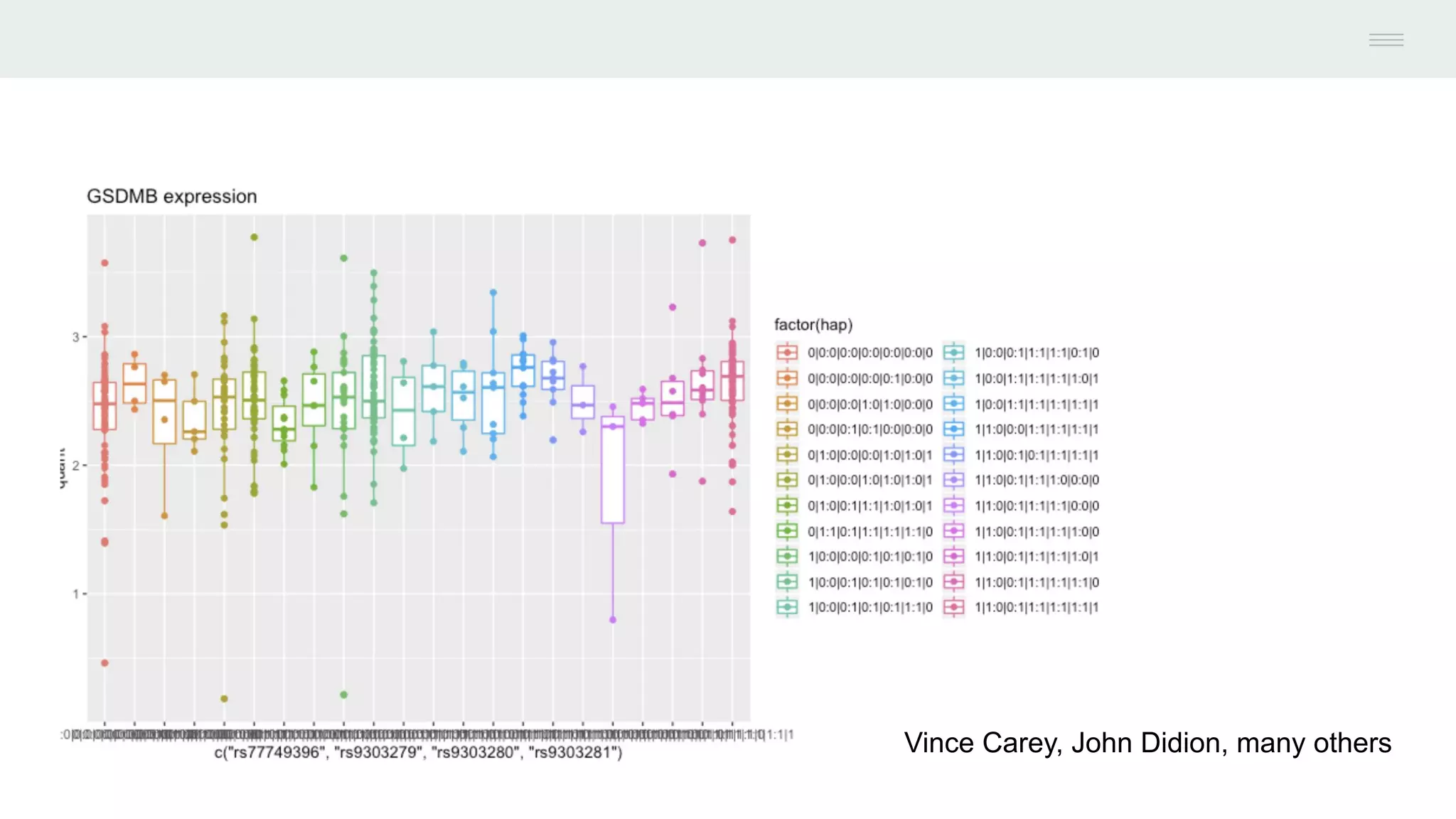
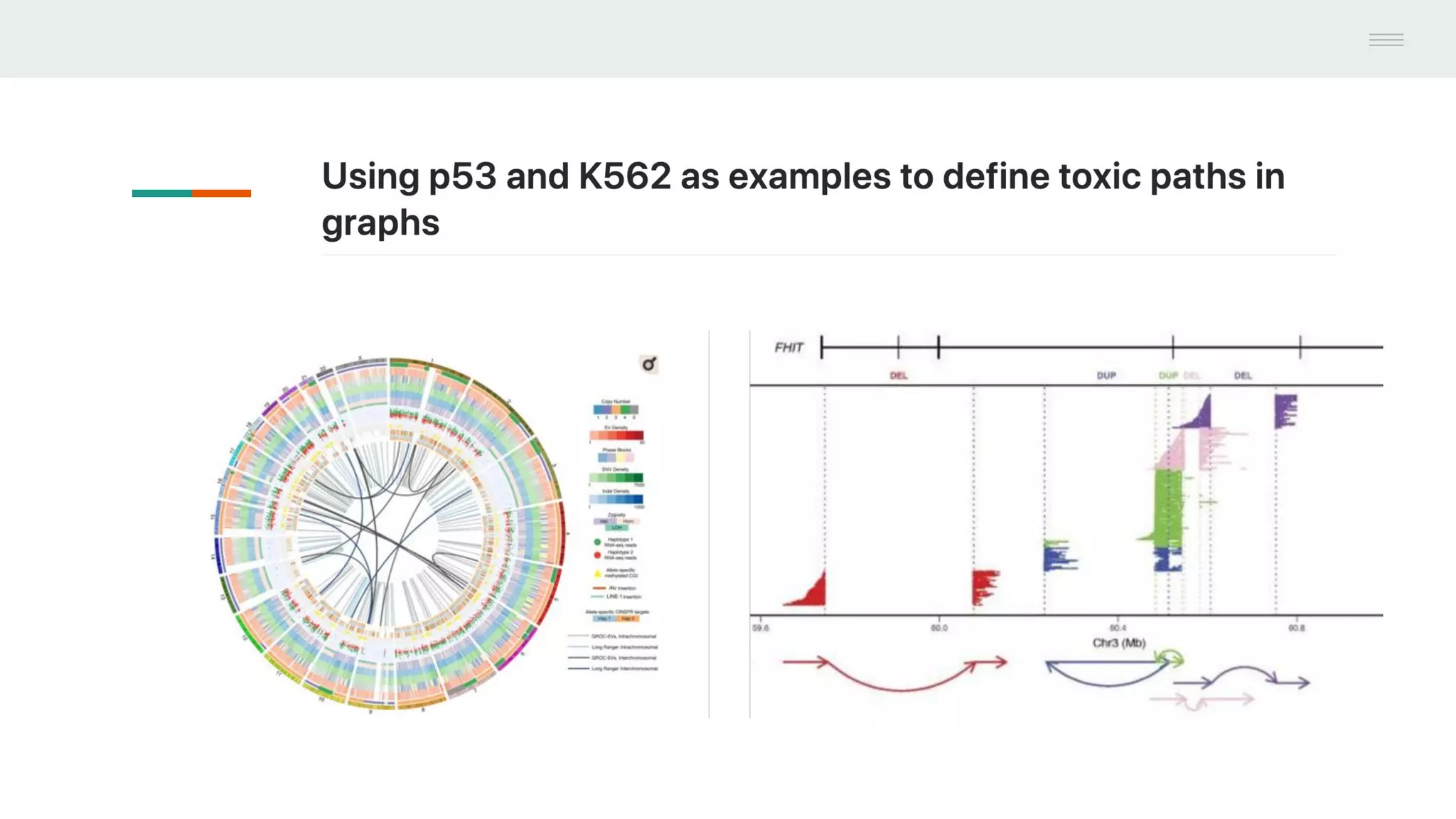
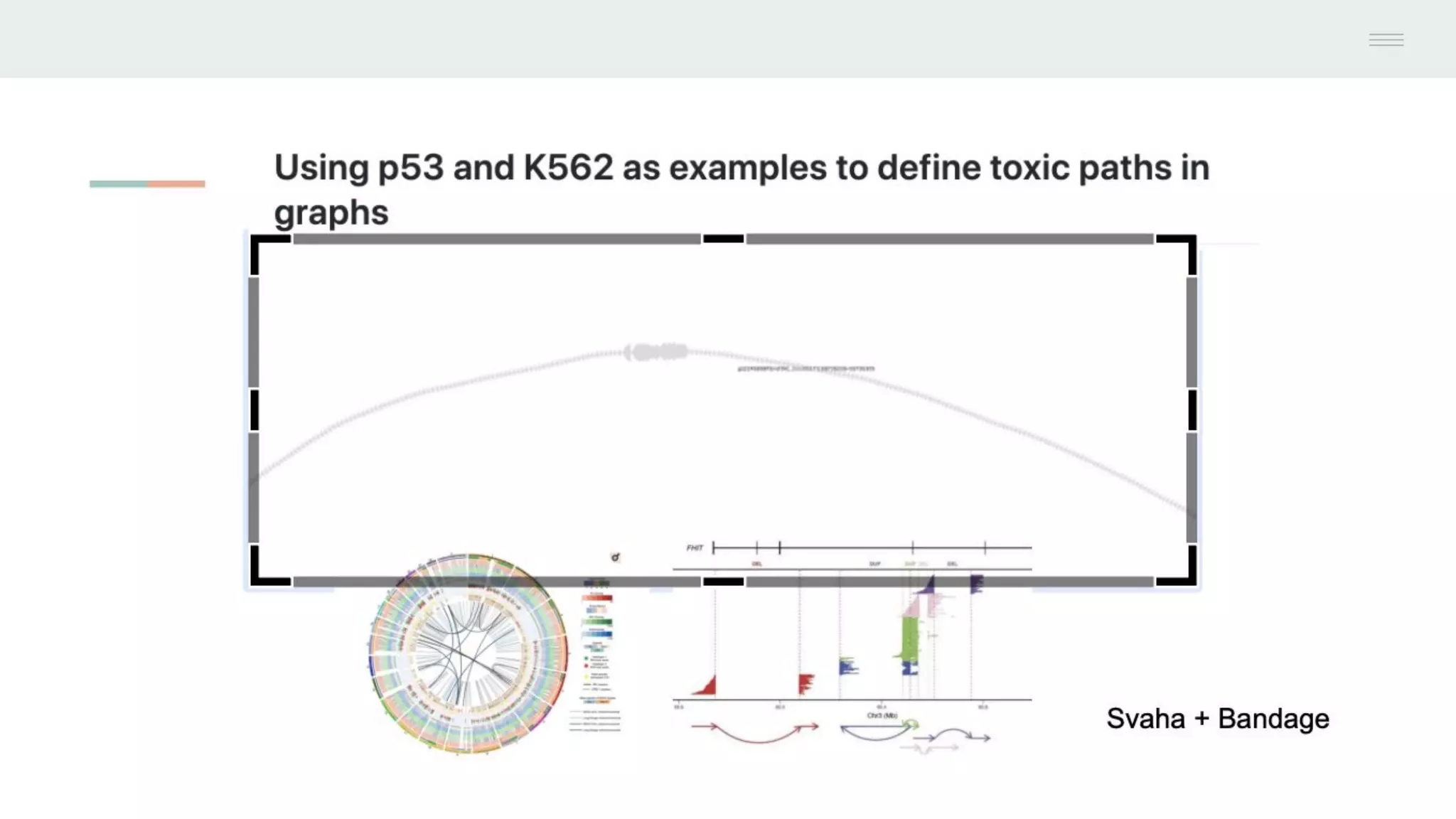



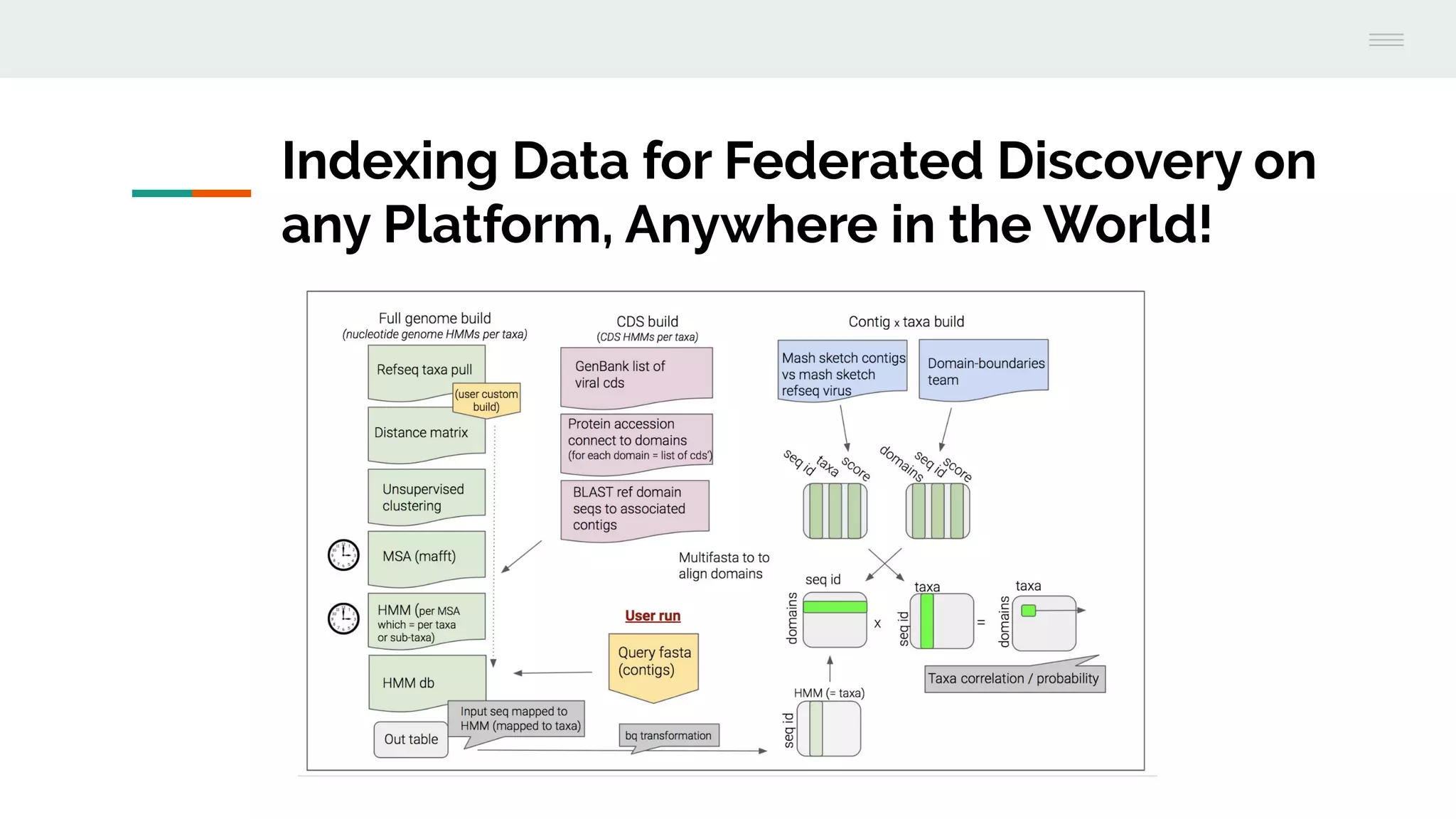
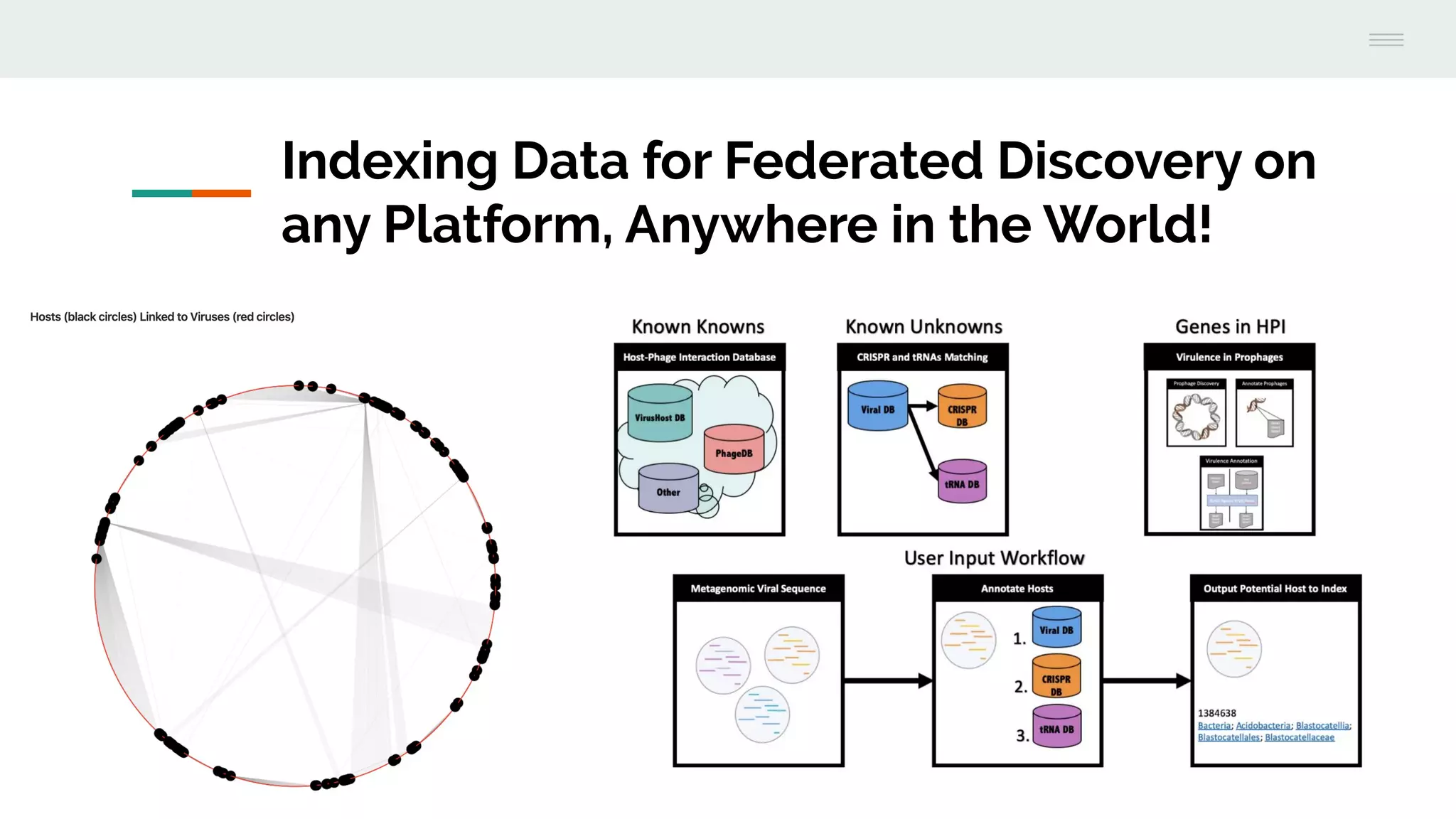



This document discusses federating genomic sequencing data across cloud platforms for discovery. It presents four topic areas as examples: 1) virus characterization and discovery through building an index of viral signatures, 2) generating systems to analyze genome graphs to compare individuals to communities, 3) annotating haplotypes and graphs to query complex disease, and 4) indexing data flexibly for federated discovery anywhere through APIs. It emphasizes that metadata is needed to contextualize data and maximize its utility for answering biological questions.












![Community Finding with Applications on Phylogenetic Networks [Extended Abstract]](https://cdn.slidesharecdn.com/ss_thumbnails/extendedabstract-190703140727-thumbnail.jpg?width=640&height=640&fit=bounds)









































































