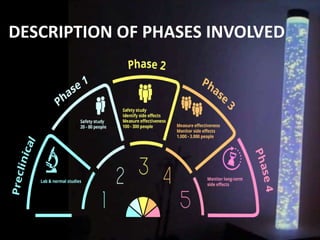
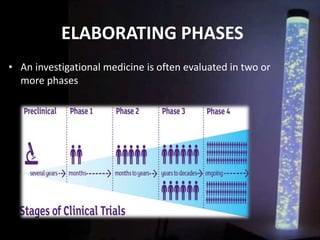
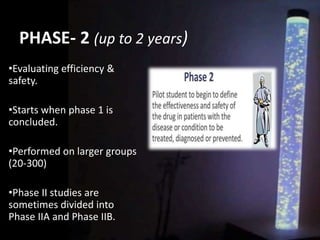
The document discusses the process of clinical trials, including the phases involved from pre-clinical to post-marketing. It describes the goals of each phase from evaluating safety and efficacy on healthy volunteers to large patient groups. Regulatory authorities ensure quality and protect subject rights through establishing legal frameworks. Pharmacists play roles in storage, supply, patient education and conducting research to improve outcomes.