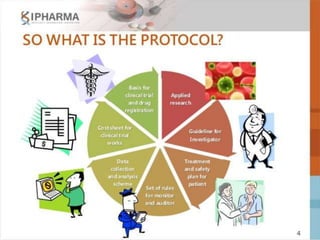
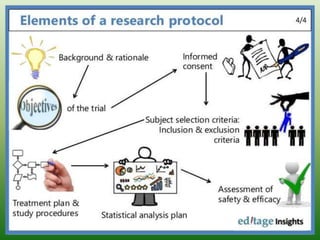
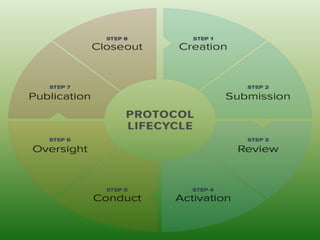
The document provides an overview of the key components that should be included when developing a clinical trial protocol. It discusses that a protocol establishes the objectives, methodology, and organization of a clinical trial to ensure safety and integrity. The document then outlines the specific sections that are typically included in a protocol according to ICH guidelines, such as title, background, objectives, study design, eligibility criteria, treatment plan, safety monitoring, statistics, and informed consent procedures. It emphasizes that a protocol provides the details necessary to properly conduct a clinical investigation.