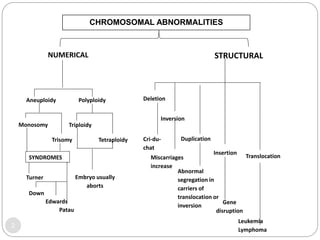
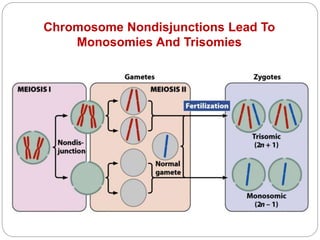
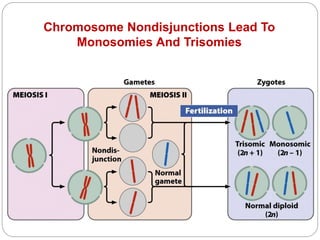
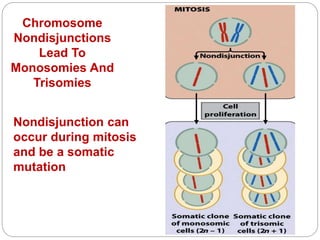
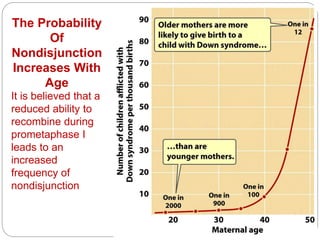
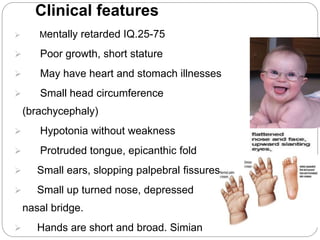
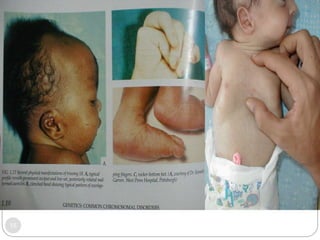
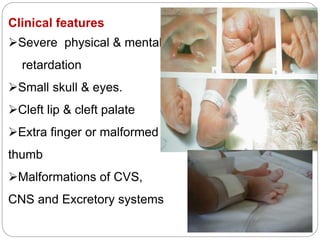
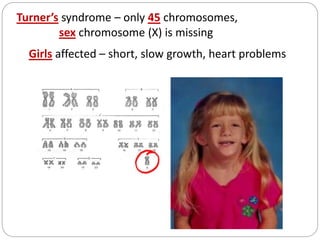
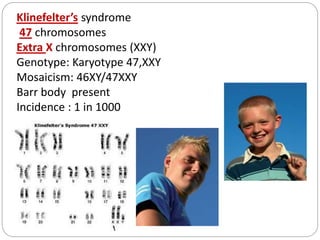
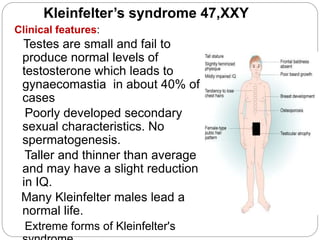
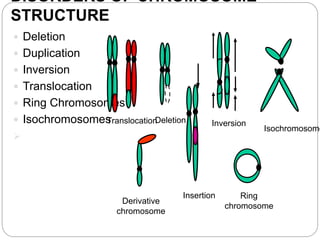
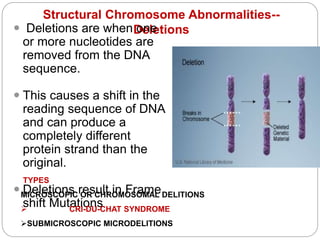
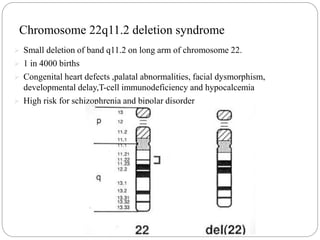
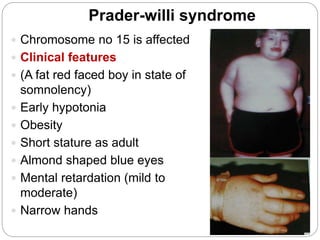
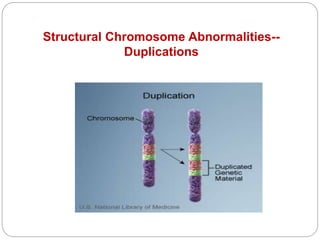
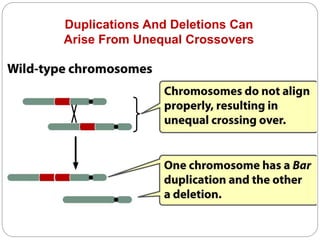
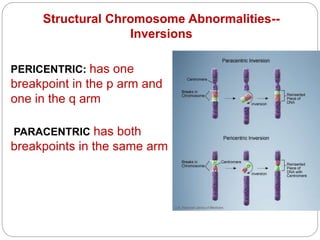
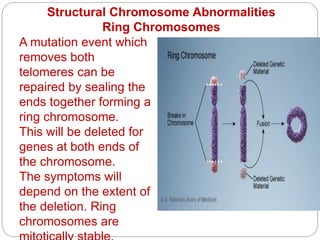
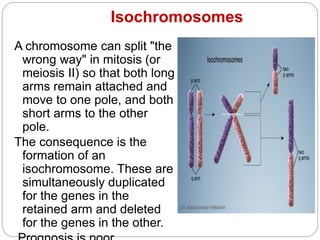
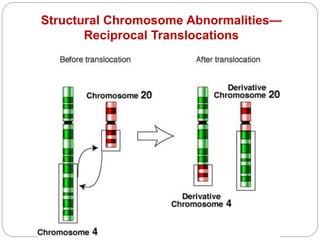
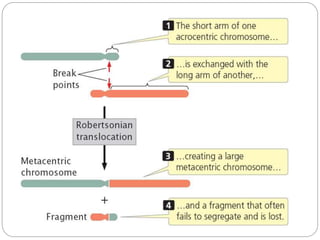
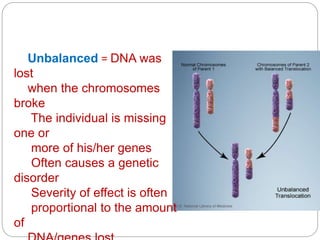
This document discusses various types of chromosome abnormalities including numerical abnormalities (involving extra or missing chromosomes) and structural abnormalities (involving changes in chromosome structure). It describes several specific chromosome disorders including Down syndrome, Edward's syndrome, Patau's syndrome, Turner syndrome, Klinefelter syndrome, and others. It explains the causes and clinical features of these conditions. It also discusses structural changes like deletions, duplications, inversions, translocations, ring chromosomes, and isochromosomes that can occur at the chromosome level.