Download to read offline
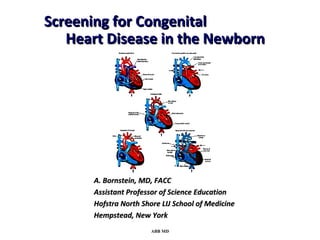
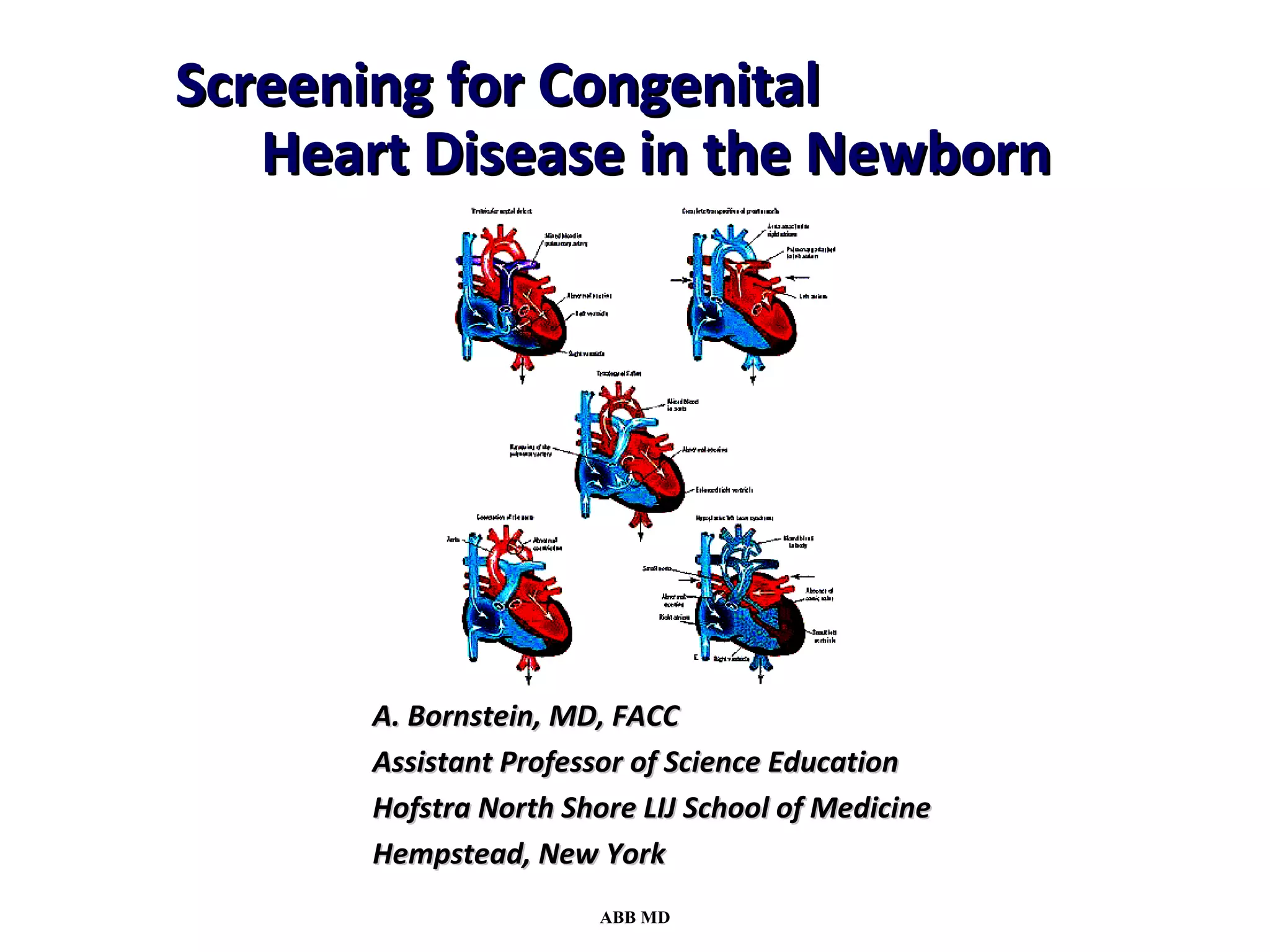


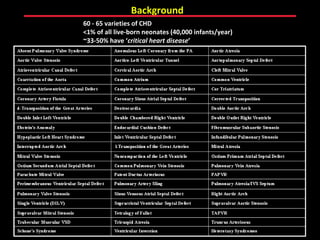
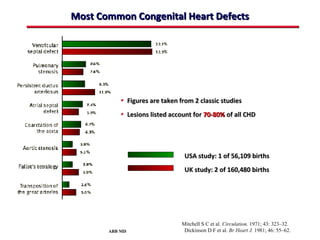
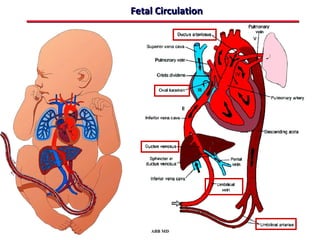
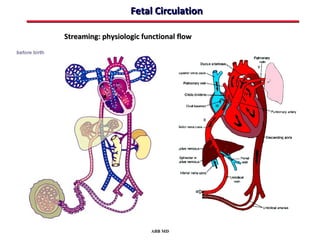
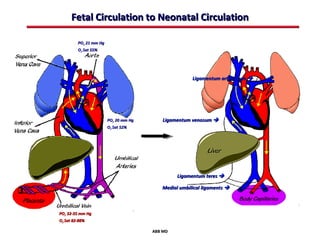
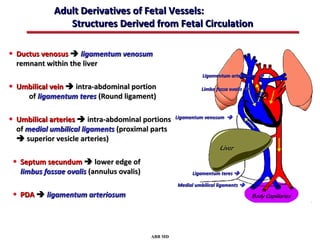
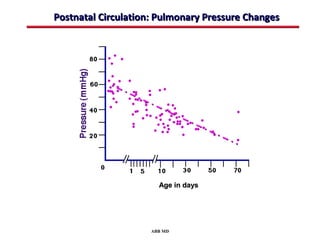
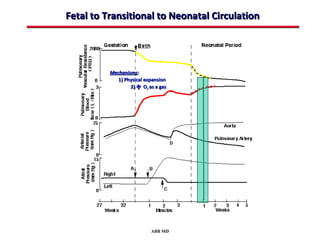
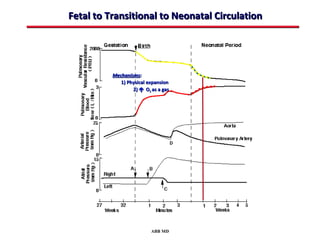
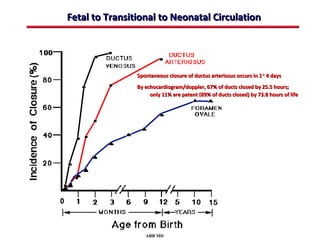
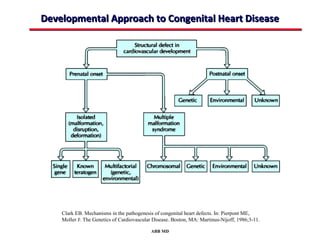



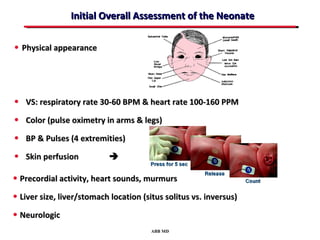
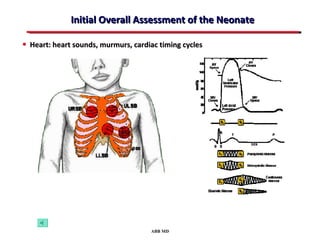
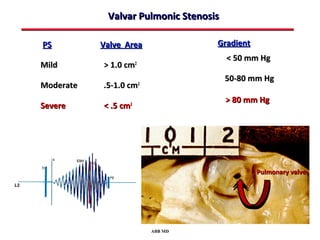
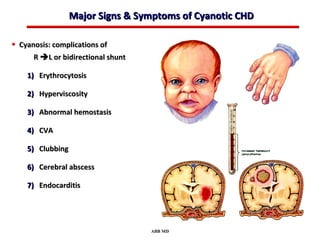
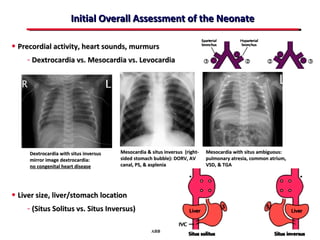
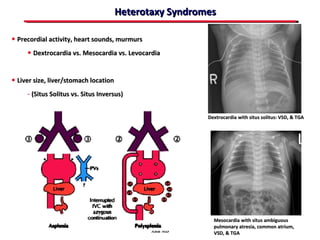
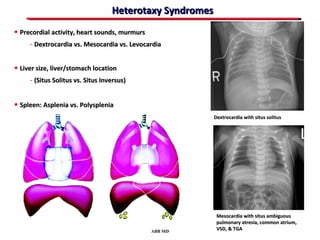
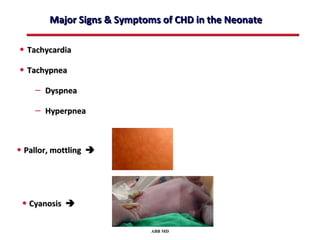


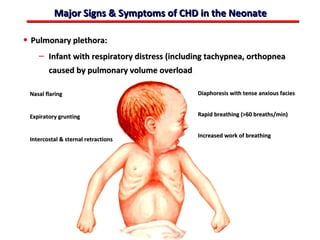


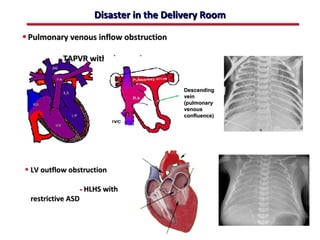
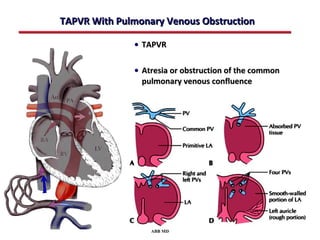
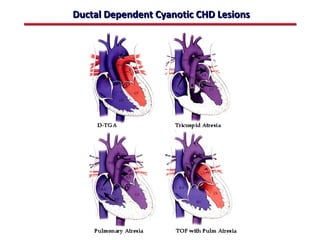
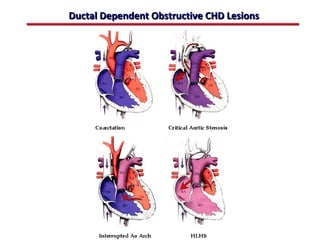
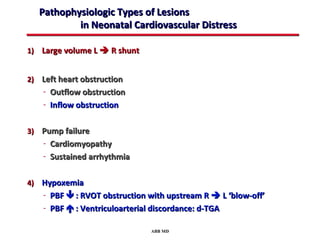

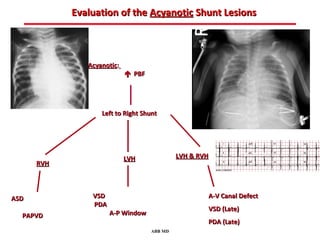
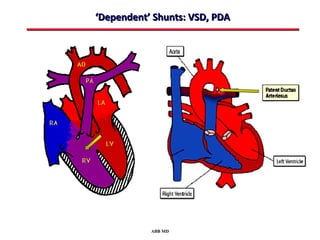
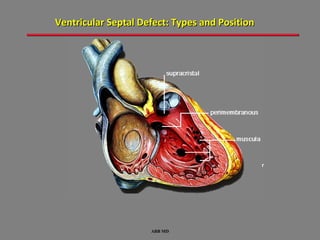
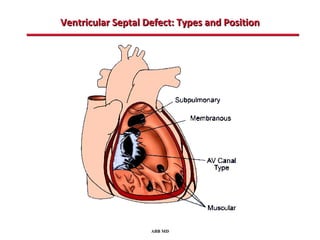
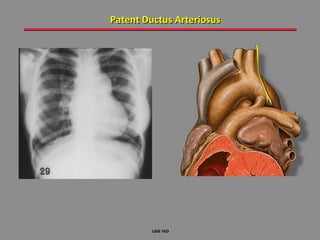
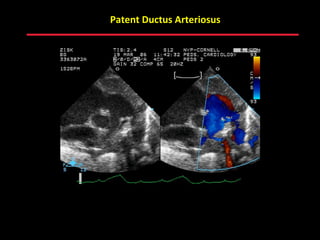
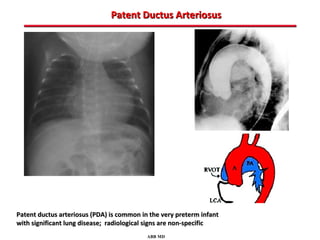
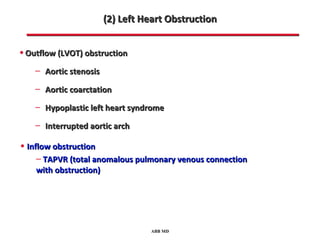


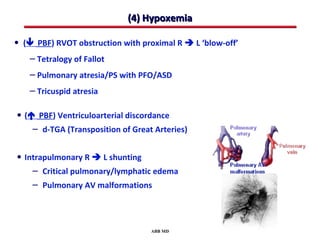


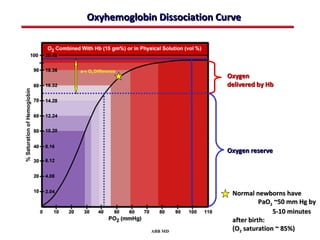
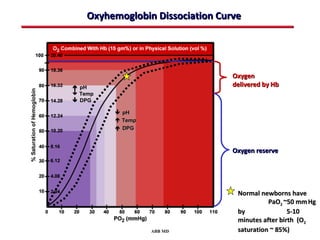
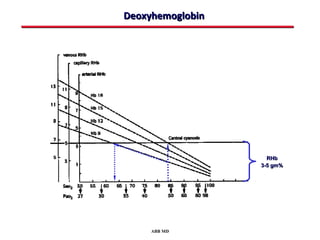
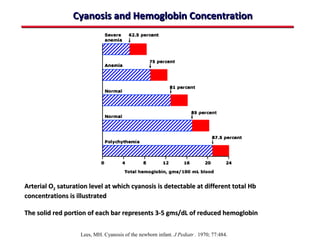





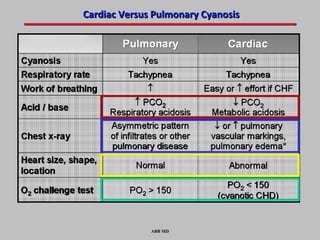
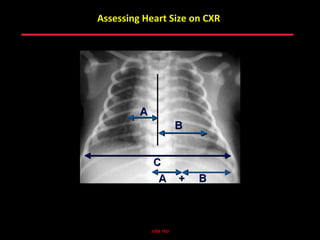
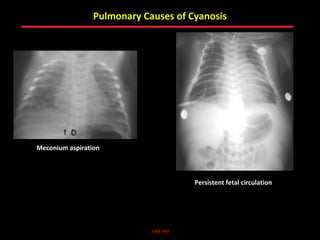
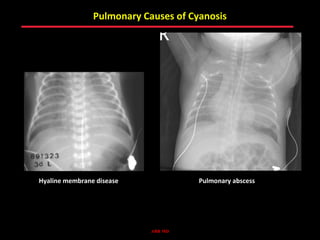
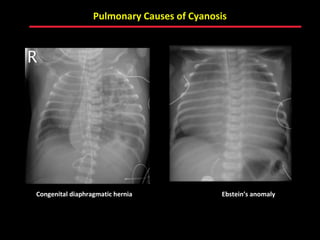
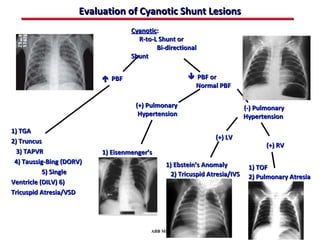

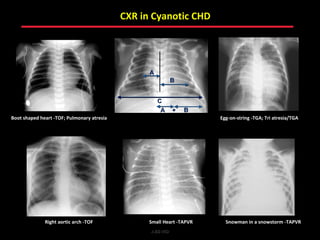
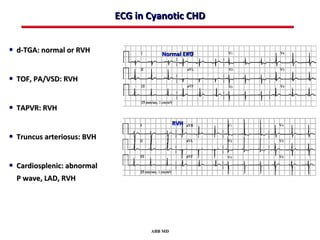
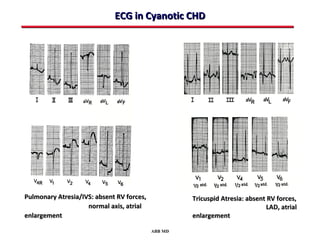
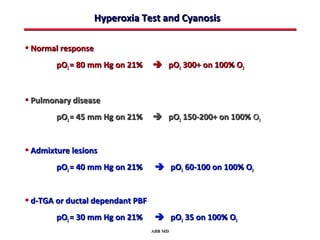
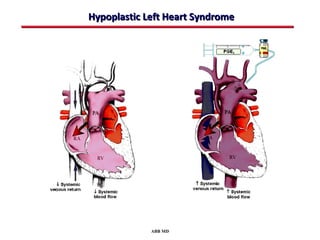

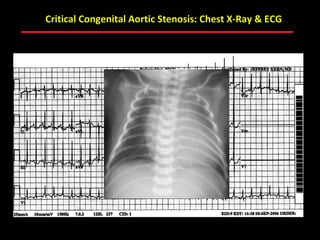
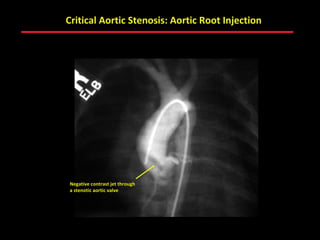
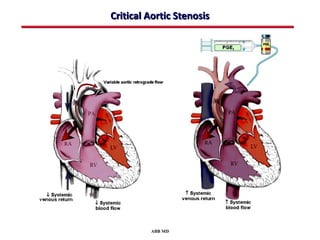

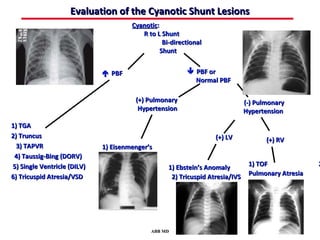


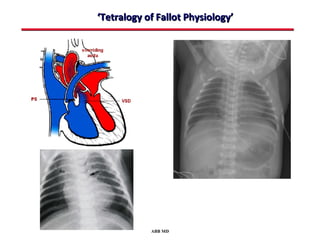

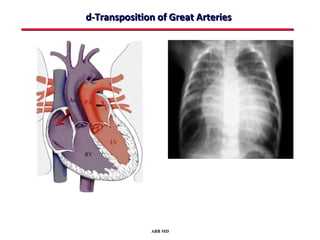
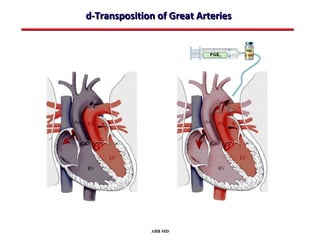






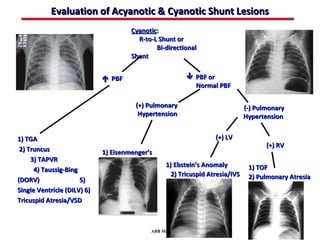
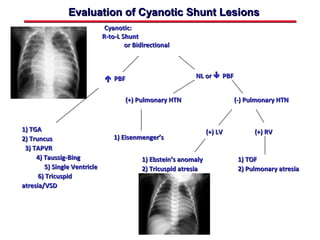
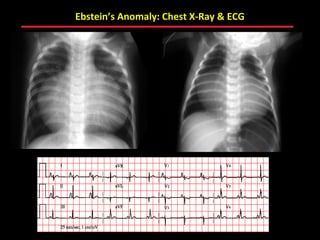
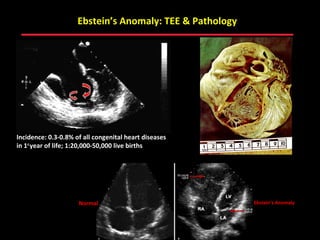
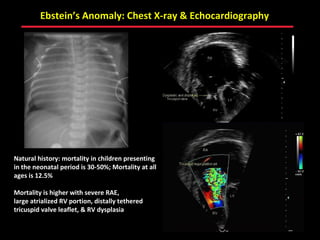
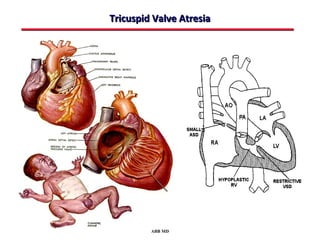

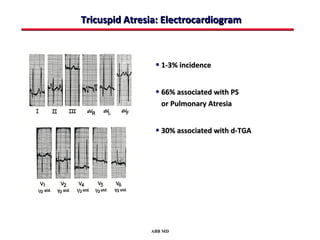
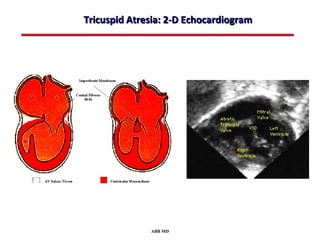
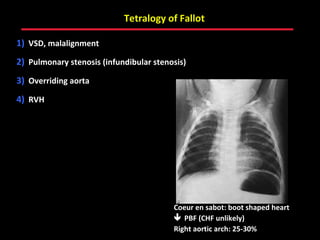
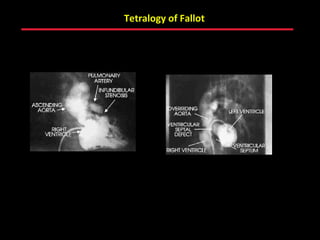
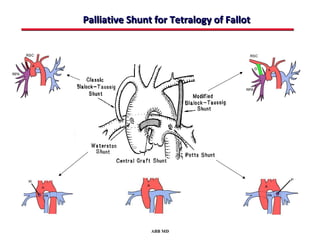
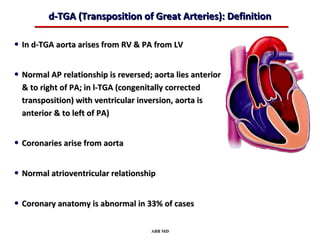


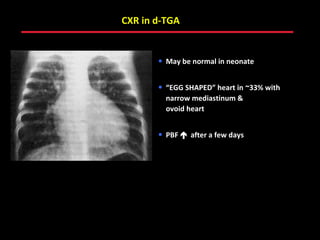
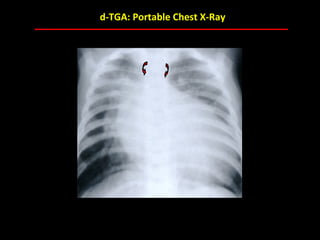
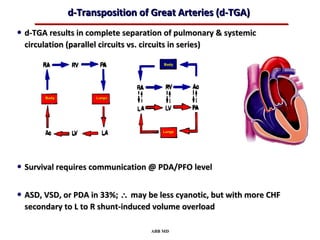


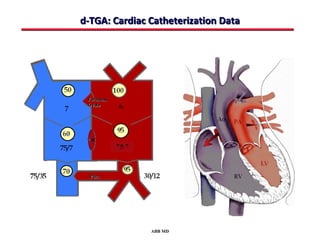
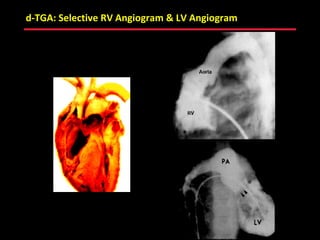
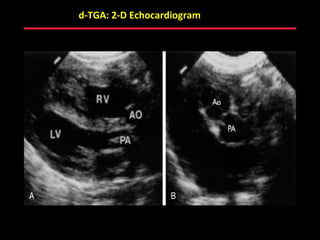
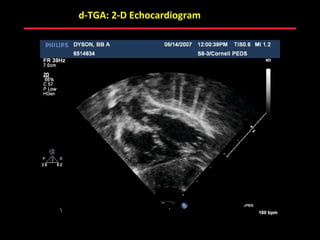
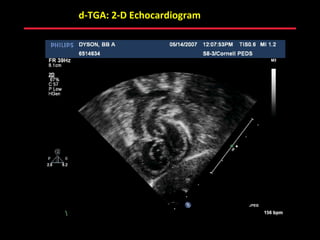
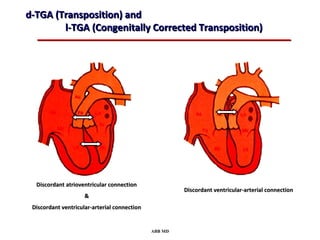


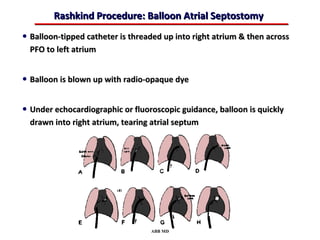
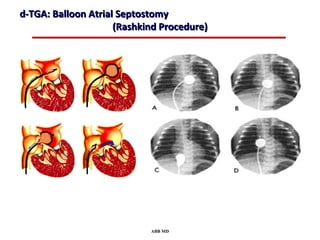
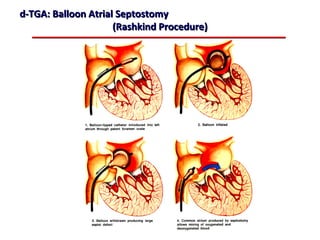










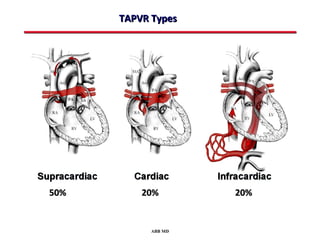
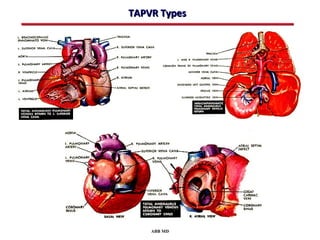
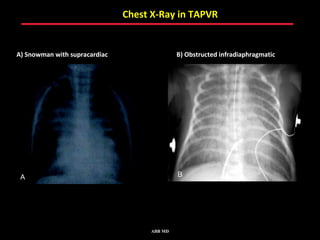
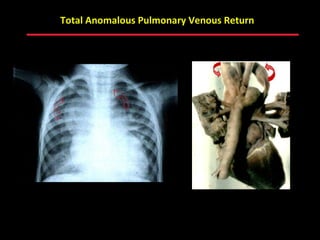
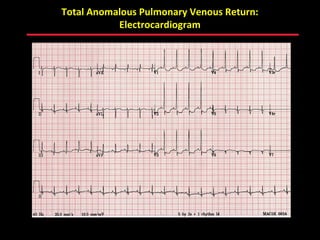
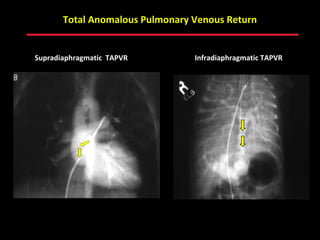
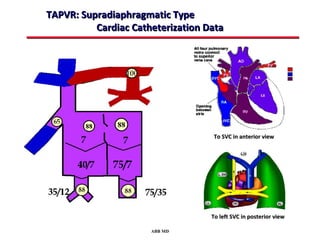
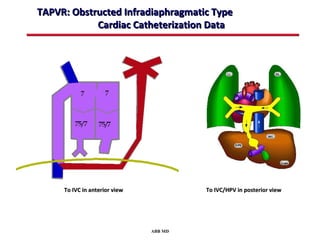

This document provides an overview of screening for congenital heart disease in newborns. It discusses the prevalence and types of congenital heart defects, the fetal circulation and how it transitions to neonatal circulation, signs and symptoms of defects seen in newborns, and approaches to evaluating newborns with suspected defects. Key topics include the pathophysiology of various defect types, how defects present in newborns, and factors affecting the detection of cyanosis. The goal is to enhance understanding of congenital heart disease and simplify the approach to management of affected newborns.





















































