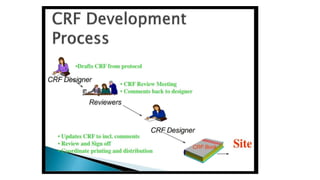
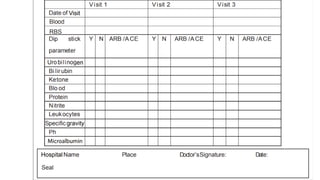
The document outlines the importance and structure of Case Report Forms (CRFs) used in clinical research, emphasizing compliance with International Council for Harmonization (ICH) guidelines. It details best practices for CRF design, advantages and disadvantages of paper-based versus electronic CRFs, and the role of CRF Completion Guidelines (CCGs) in ensuring accurate data collection. Additionally, it discusses document retention responsibilities and the need for clear guidelines for investigators to maintain the integrity of trial records.