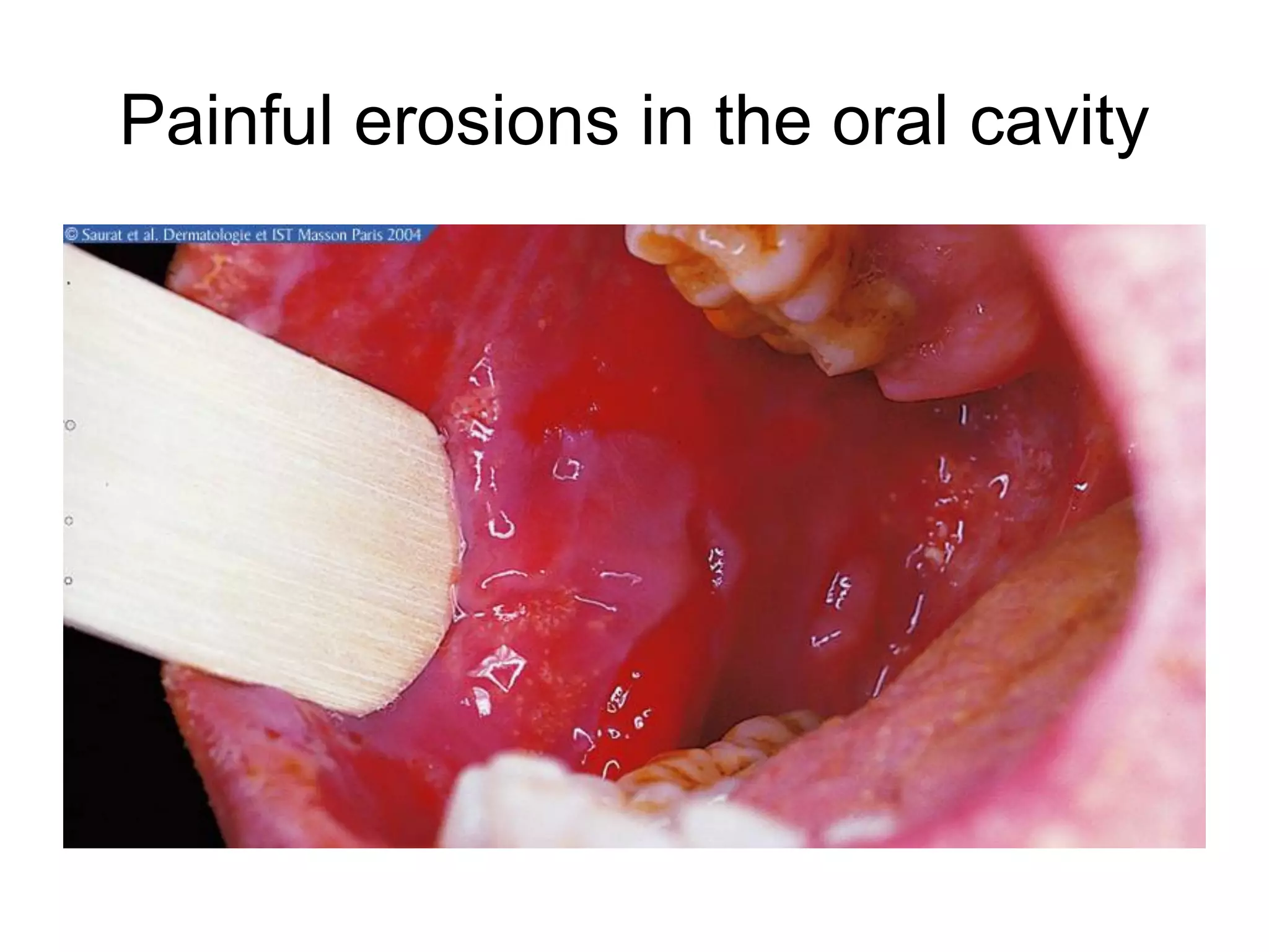
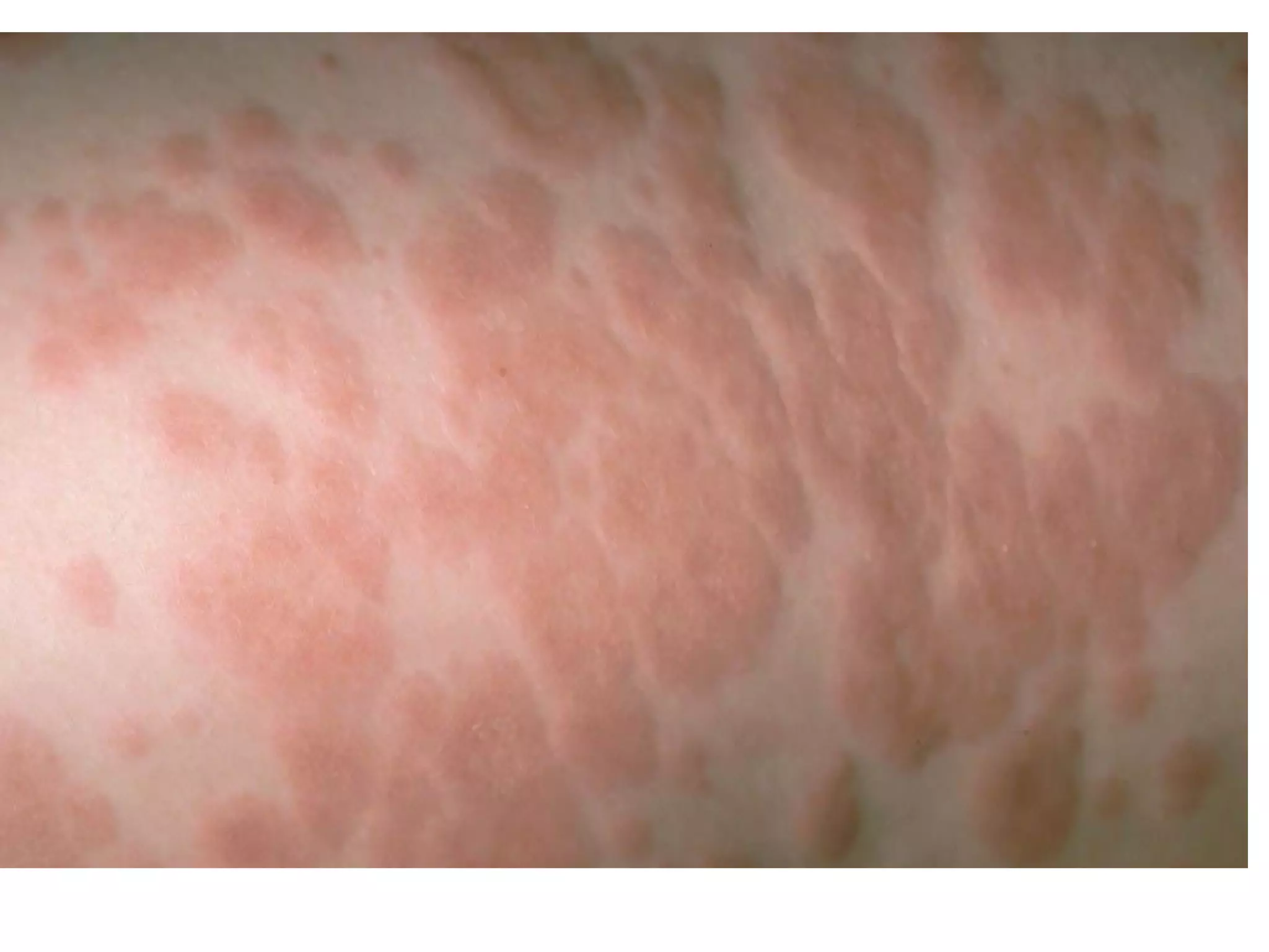
This document discusses bullous dermatoses and provides classifications and details on various autoimmune bullous diseases. It begins by classifying bullous dermatoses as either acquired (autoimmune, infectious, toxic) or hereditary (Epidermolysis Bullosa, ichtyosis bullosa). It then provides details on the structure of the epidermis and dermis, specifically focusing on the proteins involved in epidermal and dermal adhesion. The remainder of the document discusses the clinical features, histopathology, immunopathology and treatment of various autoimmune bullous diseases including Pemphigus vulgaris, Paraneoplastic Pemphigus, Bullous Pemphigoid,



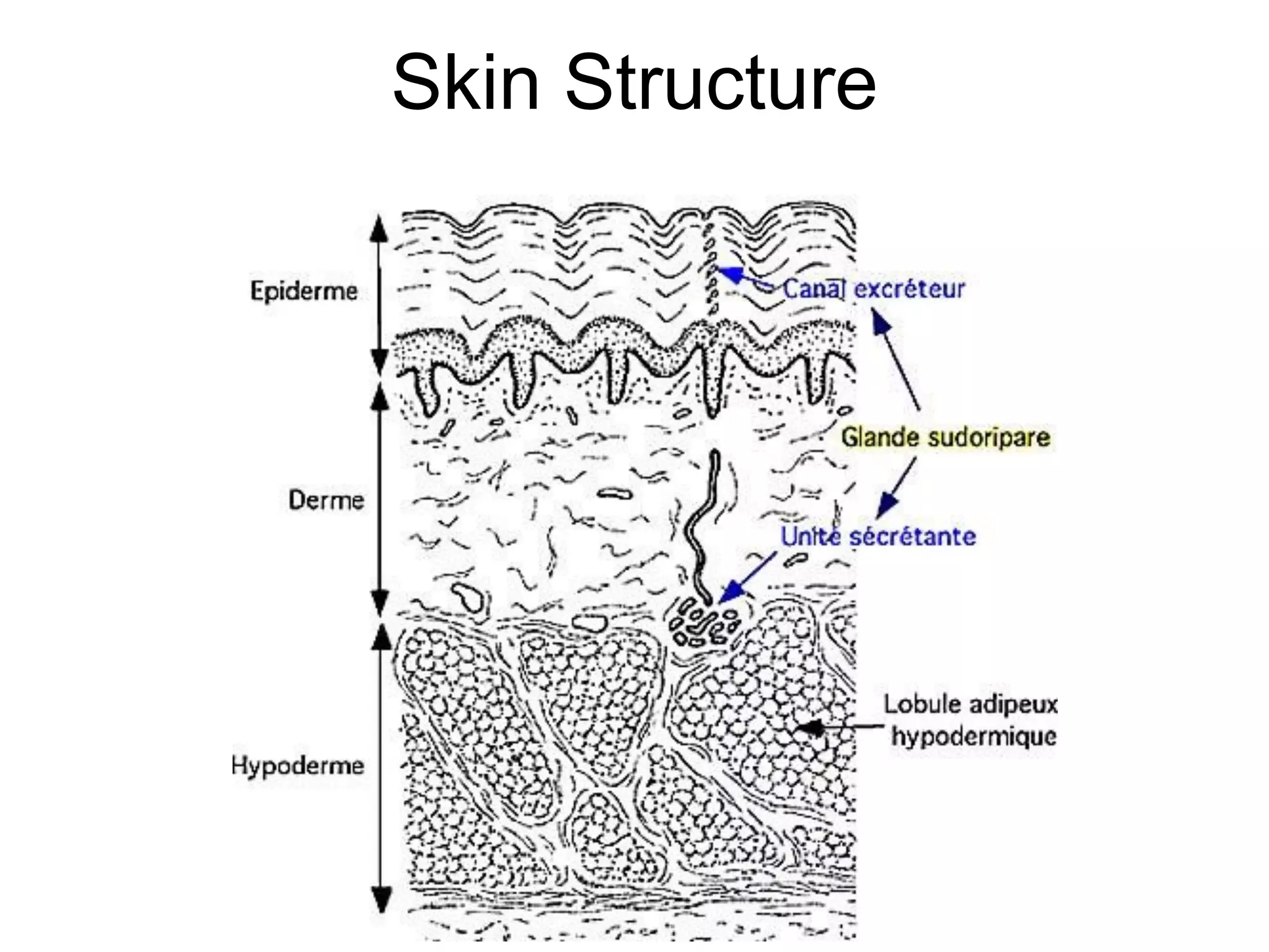




![Epidermal and Epidermal-
Dermal Cohesion
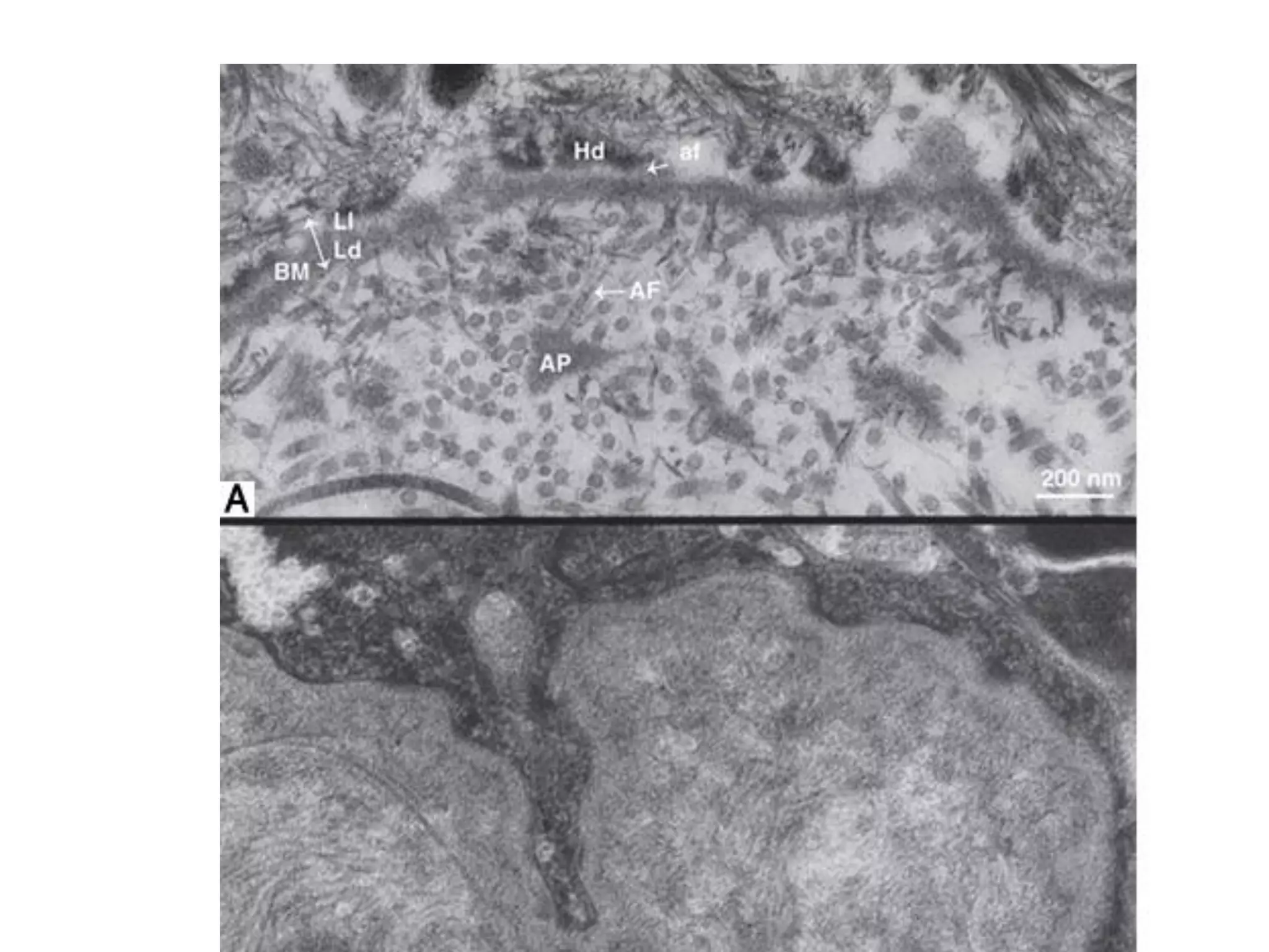
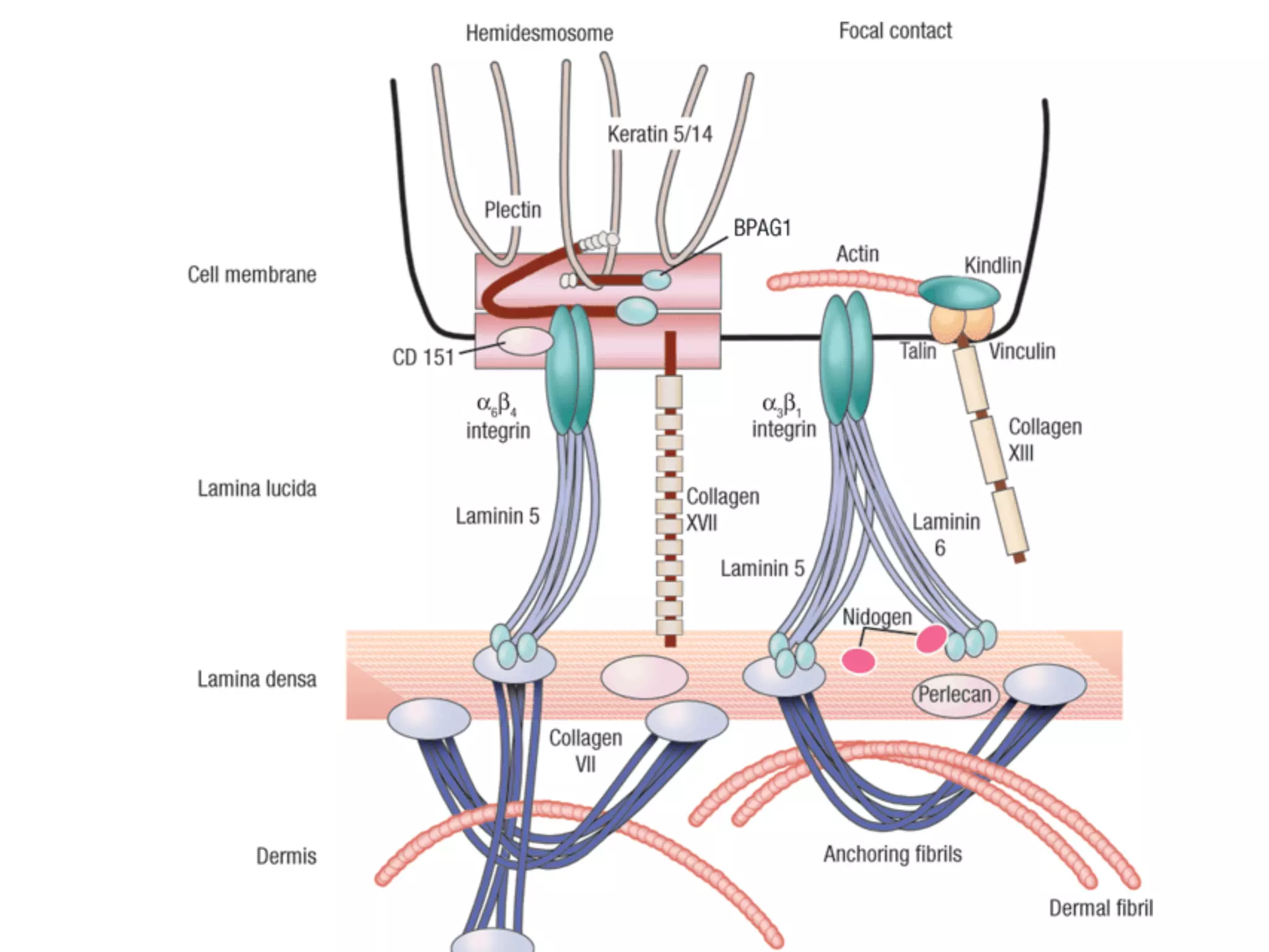
• The hemidesmosomal components comprise:
1. Cytoplasmic proteins [ plakin homologues (plectine,
BPAG1)]
2. Trasmembrane components [integrins, and collagenous
transmembrane (BPAG2)].
• All basement membranes contain collagen IV, laminins,
nidogens, and perlecan.
• Anchoring fibrils ( to the dermis) are composed mainly of
collagen VII](https://image.slidesharecdn.com/bullouslesions-230212155330-1591c28a/75/Bullous-lesions-pdf-9-2048.jpg)