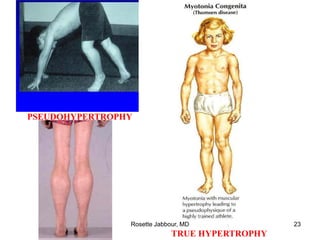
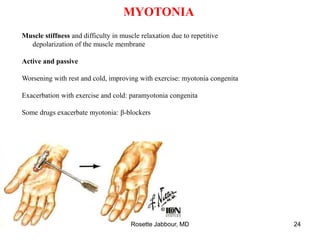
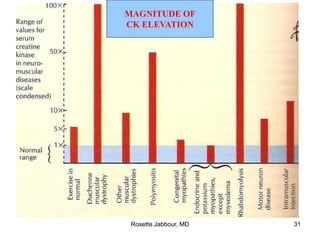
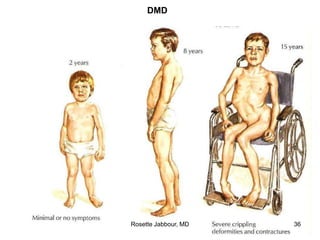
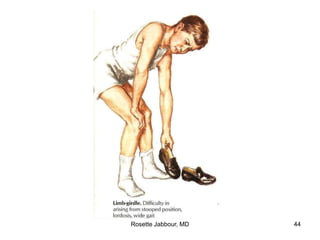
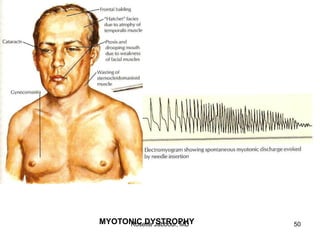
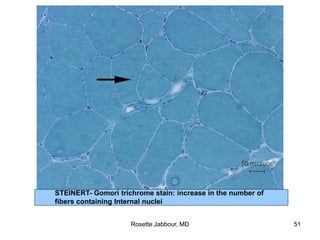
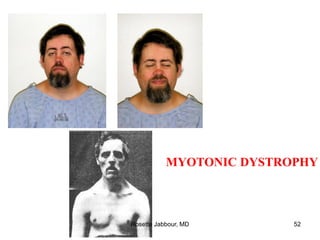
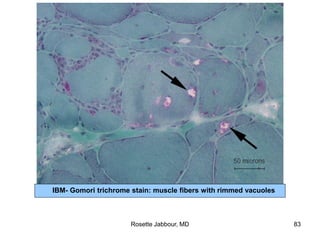
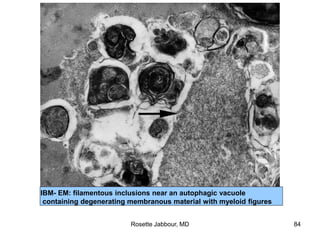
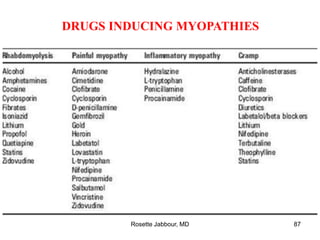
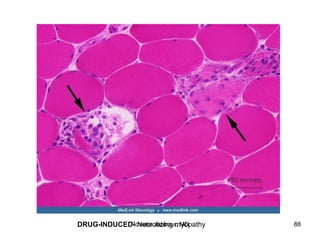
This document provides information on muscle diseases and myopathies. It discusses the classification, diagnosis, clinical features, histopathology, and genetics of various muscle disorders. Some key points include:
- Muscle diseases can be hereditary or acquired and affect skeletal muscle structure or function. Around 1/1000 people have a primary muscle disease.
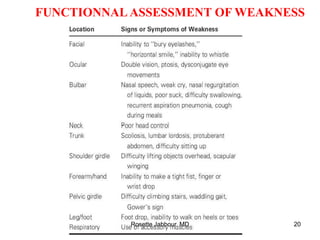
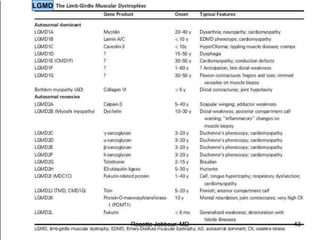
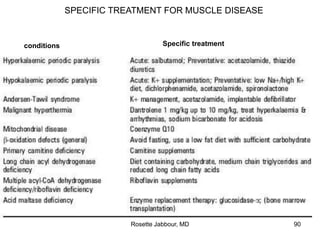
- Diagnosis involves confirming a myogenic syndrome, determining the etiology through various tests, and ruling out neurogenic or other causes.
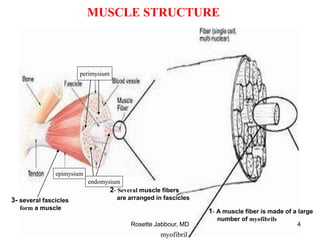
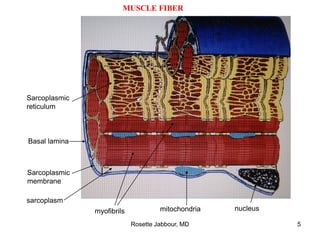
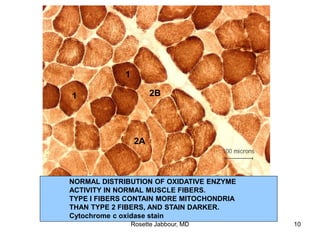
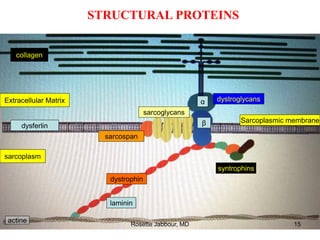
- Muscle is composed of fascicles of muscle fibers which contain myofibrils and other structures. There are different types of muscle fibers.
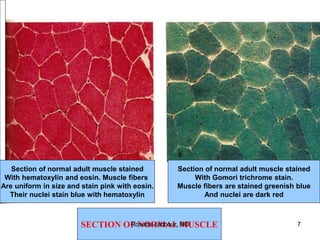
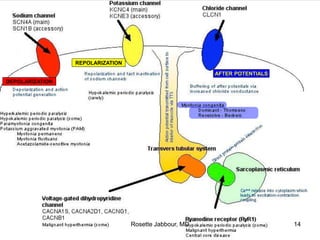
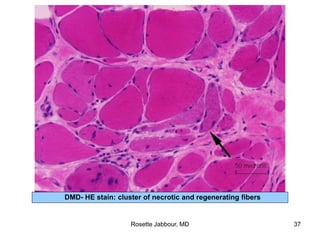
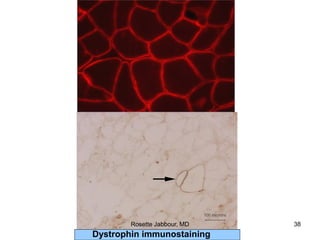
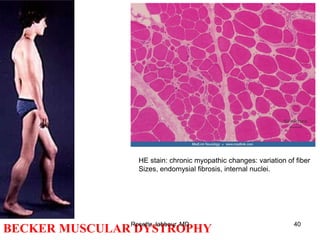
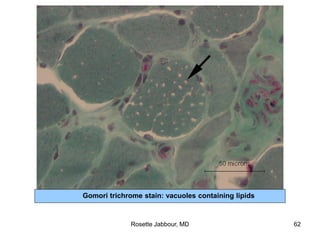
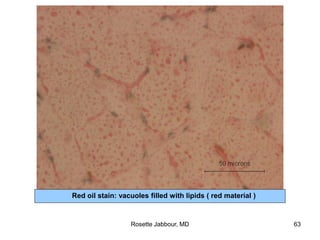
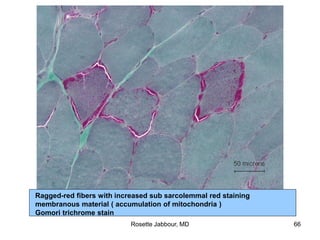
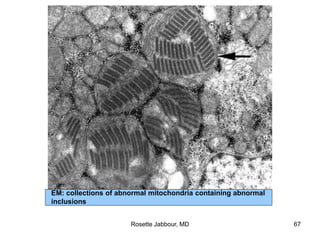
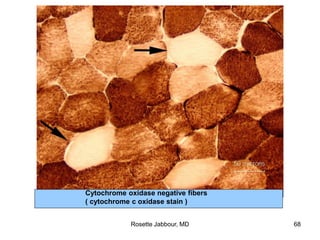
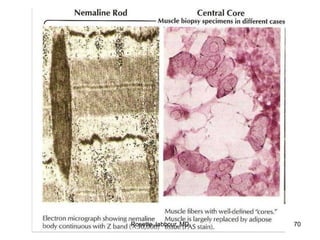
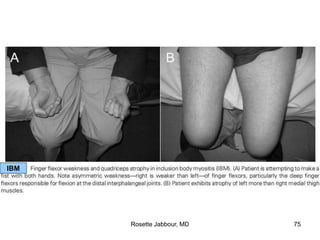
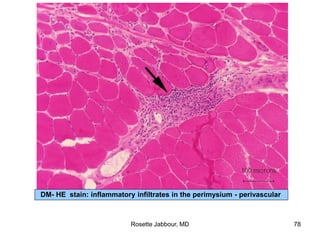
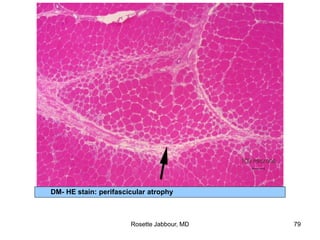
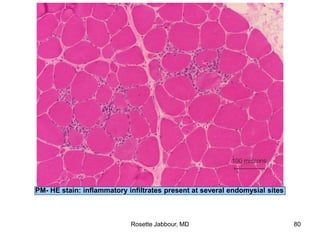
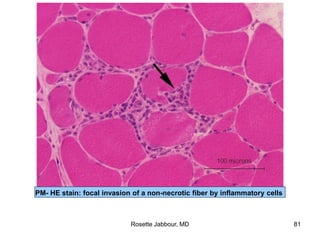
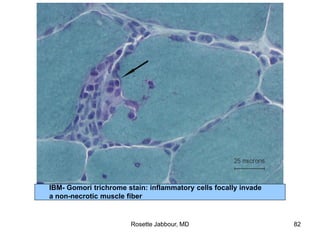
- Histopathology can identify features of muscle disease like fiber atrophy, necrosis, regeneration