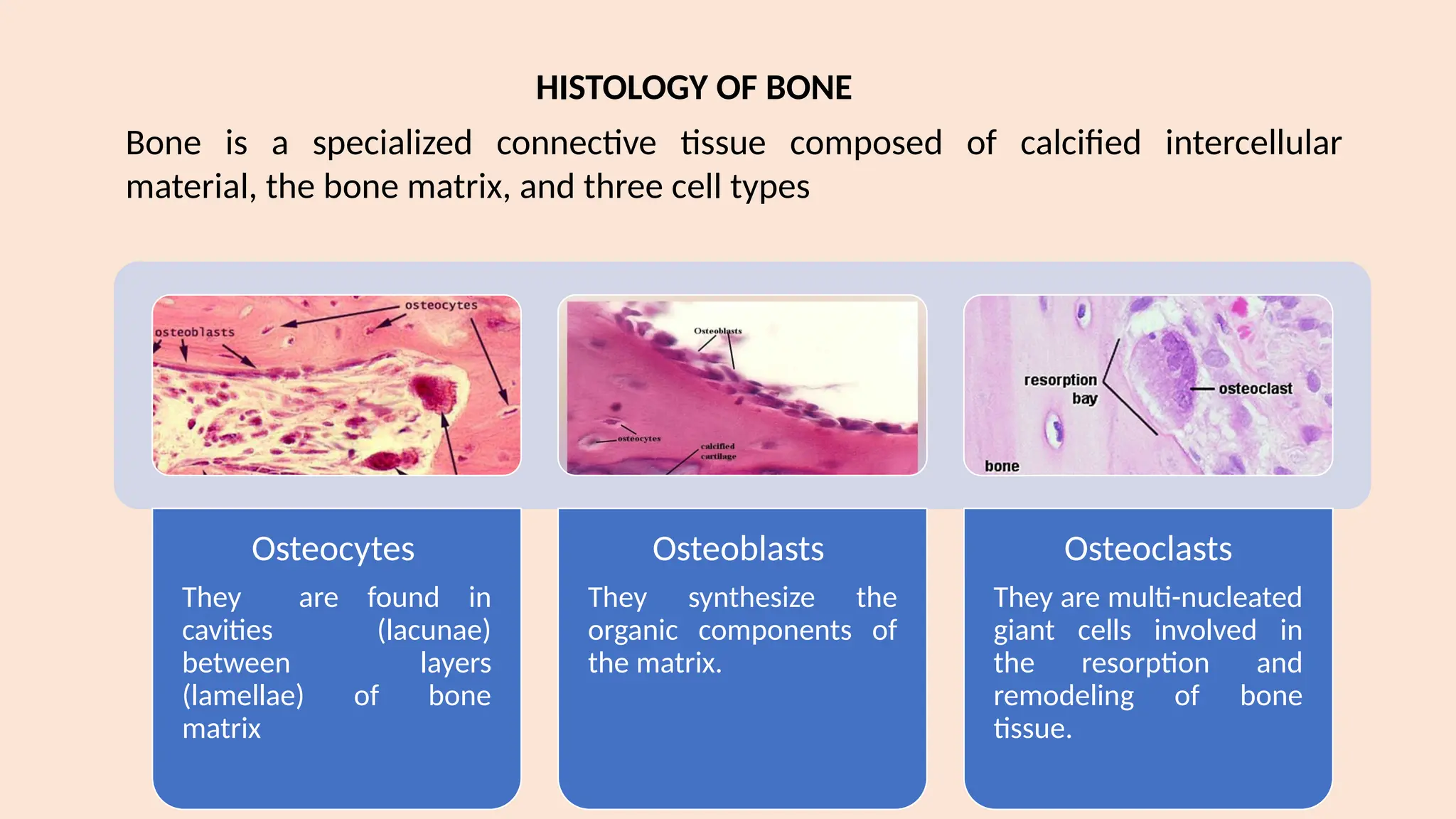
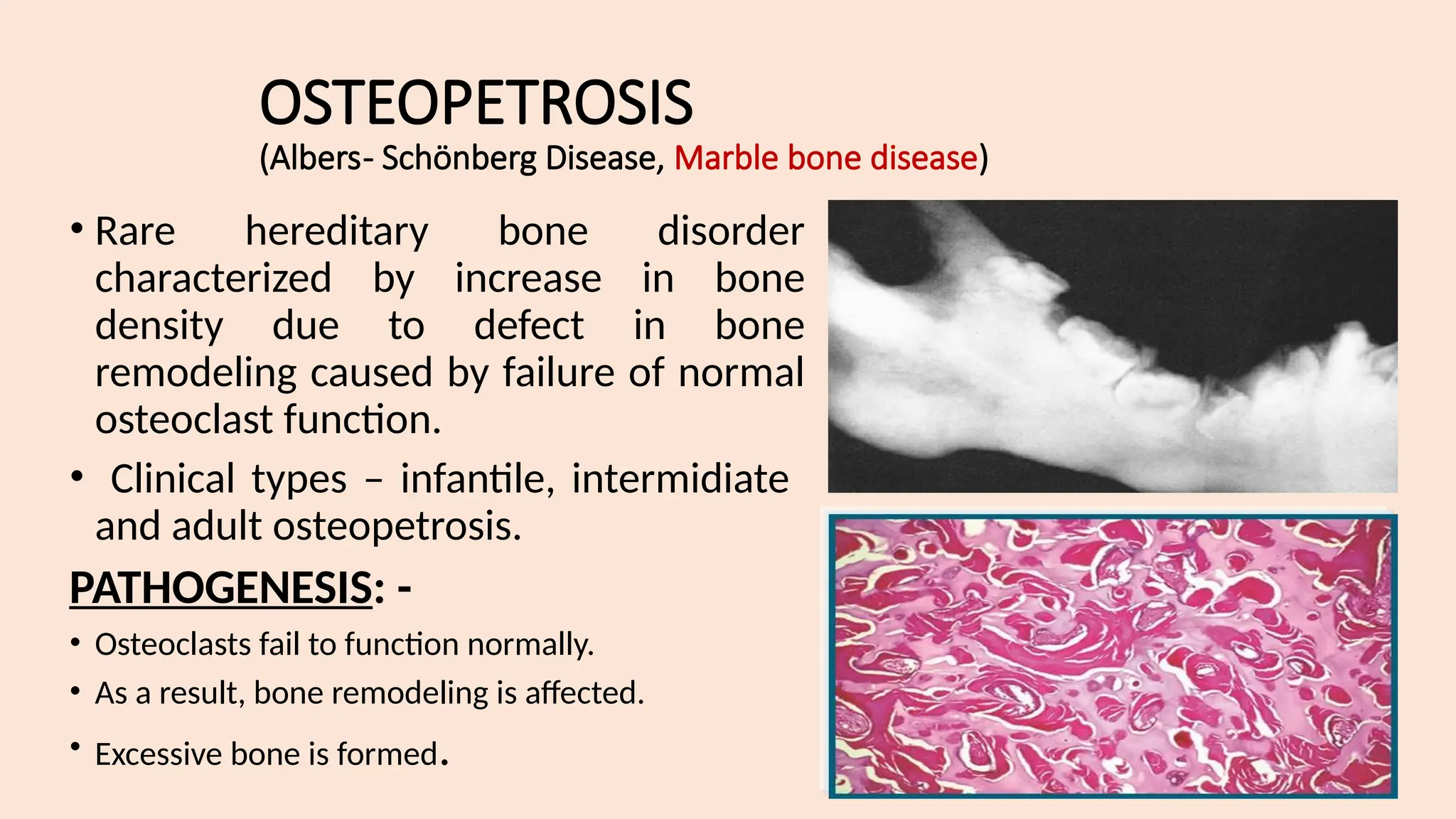
The lecture by Dr. Diana Prem covers bone and joint diseases, emphasizing the types of cells involved in bone structure, the process of bone remodeling, and various skeletal system diseases, including genetic, endocrinal, idiopathic, reactive, fibro-osseous, inflammatory, and neoplastic diseases. Key conditions discussed include fibrous dysplasia, cherubism, cemento-osseous dysplasia, and Paget's disease, detailing their clinical and radiographic features as well as oral manifestations and treatment options. The lecture highlights the importance of a multidisciplinary approach for managing these conditions and understanding their pathology.