Downloaded 1,999 times




























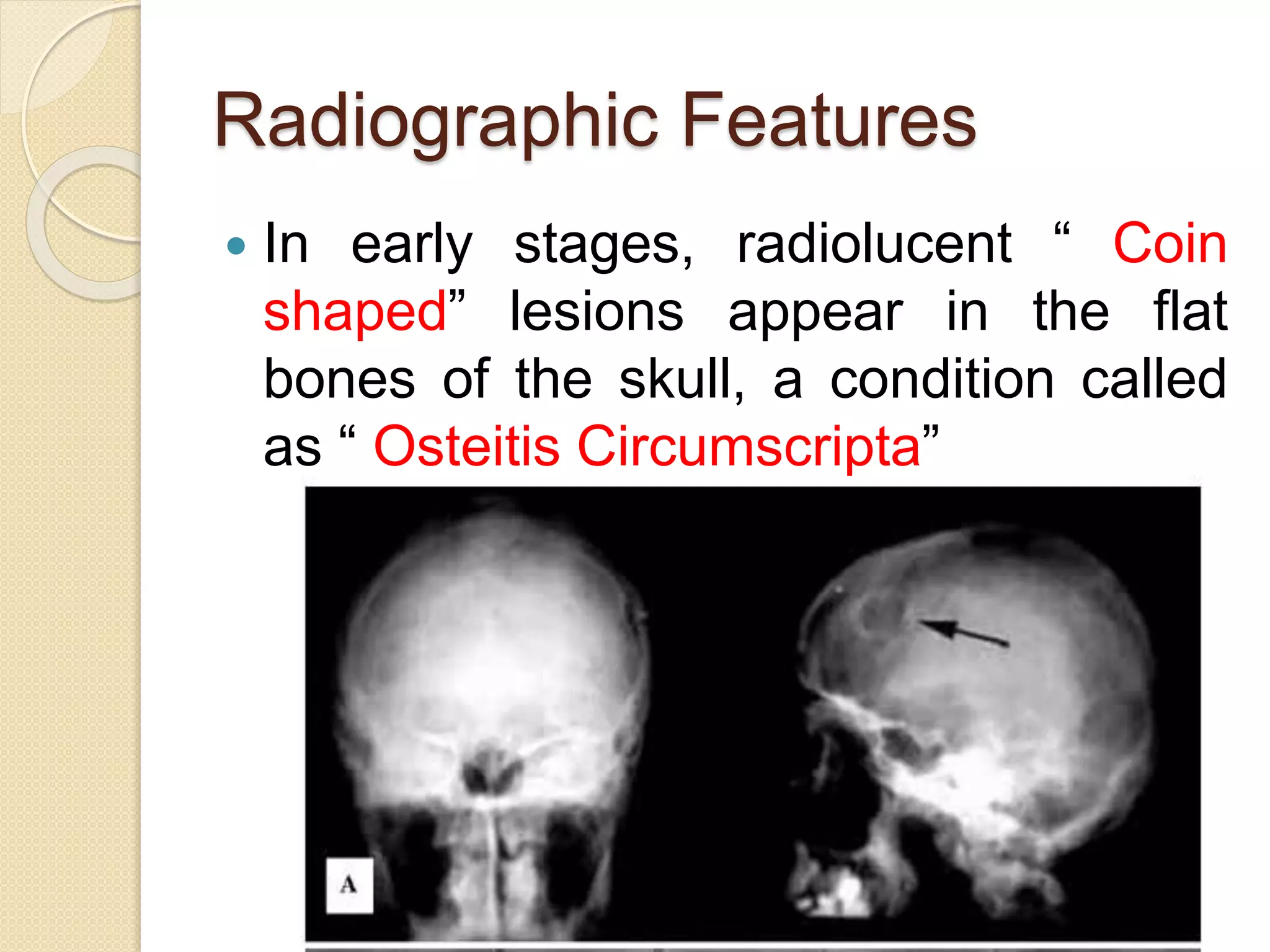
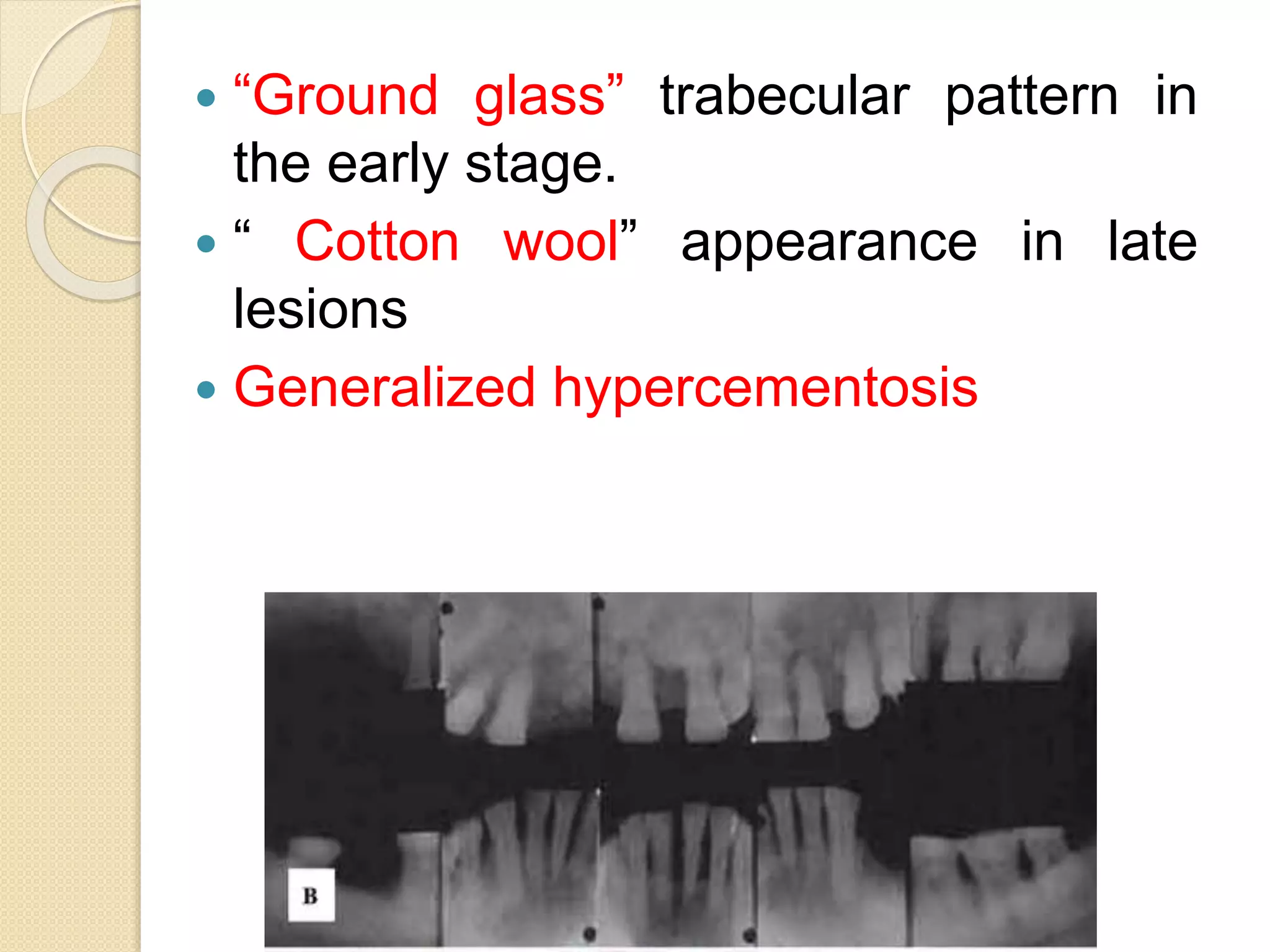
























































This document summarizes fibro-osseous lesions (FOLs), which are characterized by the replacement of bone by a benign connective tissue matrix displaying varying degrees of mineralization. FOLs include fibrotic dysplasia, cemental lesions arising from the periodontal ligament, and fibro-osseous neoplasms. Fibrotic dysplasia is caused by a GNAS1 gene mutation and can be monostotic (single bone) or polyostotic (multiple bones). Polyostotic fibrotic dysplasia can occur with skin pigmentation and endocrine disorders. Treatment depends on symptoms and may include observation, medication such as bisphosphonates, surgical remodeling, or radical excision.



































































