Download as PDF, PPTX










































































































![104
Clinical presentation
Symptoms can be divided into 2 groups.
Symptoms due to local mass effects of the tumor
Symptoms depend on the size of the intracranial tumor.
Headaches and visual field defects are the most common
symptoms. Visual field defects depend on which part of the
optic nerve pathway is compressed.
The most common manifestation is a bitemporal hemianopsia
due to pressure on the optic chiasm.
Tumor damage to the pituitary stalk might cause
hyperprolactinemia due to loss of inhibitory regulation of
prolactin secretion by the hypothalamus. Damage to normal
pituitary tissue can cause deficiencies of glucocorticoids, sex
steroids, and thyroid hormone.
Loss of end organ hormones is due to diminished anterior
pituitary secretion of corticotropin (ie, adrenocorticotropic
hormone [ACTH]), gonadotropins (eg, luteinizing hormone
[LH], follicle-stimulating hormone [FSH]), and thyrotropin
(ie, thyroid-stimulating hormone [TSH]).
Symptoms due to excess of GH/IGF-I
Soft tissue swelling and enlargement of extremities
Increase in ring and/or shoe size
Hyperhidrosis
Coarsening of facial features
Prognathism
Macroglossia](https://image.slidesharecdn.com/2bf2eb75-f34e-4c8f-9abe-4b3fc5b9d875-161028110507/85/23-BONE-PATHOLOGIES-LAMBERT-109-320.jpg)












































![150
the cause, with disturbances in the normal separative process.61
It is widely
considered to be a developmental (hamartomatous) lesion. Although it is
usually classified as a nonneoplastic disorder, some examples show
neoplastic-like clinical features.62
With the occurrence of polyostotic
cherubism it strongly suggested a fundamental genetic defect of embryonic
origin. The probable mode of inheritance is highly indicative of an
autosomal dominant trait in both the monostotic and polyostotic varieties
of the condition.63
The disease is caused by a somatic mutation of GNAS1 gene
(guanine nucleotide-binding protein, alpha-stimulating activity polypeptide
1; chromosome 20). There is a so-called gain of function mutation
resulting in an increased hyper-function of osteoblasts, melanocytes &
endocrine cells.There is also an increase in IL-6-induced osteoclastic bone
resorption. Due to postzygotic mutation in the GNAS-1 gene, mutation
occurs in undifferentiated stem cells of osteoblasts, melanocytes &
endocrine cells. The progeny of the mutated cell will carry the mutation
and express the mutated gene. The clinical expression is depending on the
size of the cell mass and where in the cell mass the mutation occurs:
a) If mutation occurs in early embryonic life, it results in multiple bone
lesions, cutaneous pigmentation & endocrine disturbances (Mc-
Cune-Albright syndrome).
b) If it occurs in later stages of normal skeletal formation, it results in
multiple bone lesions [polyostotic (Jaffe type)].
c) If it occurs during postnatal life, confined to one bone, results in FD
of a single bone (monostotic fibrous dysplasia).](https://image.slidesharecdn.com/2bf2eb75-f34e-4c8f-9abe-4b3fc5b9d875-161028110507/85/23-BONE-PATHOLOGIES-LAMBERT-155-320.jpg)



















































![204
Rarely, lesions that are unusually large and painful occur in
inaccessible sites or involve vital structures. They require radiation
(3-6 Gy [300-600 rad]).
Localized skin disease is best treated with moderate-to-potent
topical steroids (eg, mometasone furoate [Elocon] cream 0.1%,
triamcinolone [Kenalog] cream 0.1%, fluocinolone [Synalar]
ointment 0.025%) or super-potent topical steroids (eg, clobetasol
propionate 0.05%).
In cases of severe cutaneous involvement, topical nitrogen mustard
(20% solution) may be used, based on its easy administration
especially in outpatient settings and freedom from adverse effects.
For single lymph node infiltration, excision is the treatment of
choice.
Regional lymph node enlargement can be treated with a short course
of systemic steroids.
Treatment-resistant nodes with sinus tracts to the skin may require
systemic chemotherapy.
Multisystem disease
Systemic chemotherapy is indicated for cases of multisystem disease
and those cases of single-system disease that are not responsive to
other treatment.
The combination of cytotoxic drugs and systemic steroids is
effective. Low-to-moderate doses of methotrexate, prednisone, and
vinblastine are used.
BONE LESIONS OF GAUCHER'S DISEASE41,42
Gaucher's disease, an autosomal recessive genetic disorder, is a
lysosomal storage disease caused by a deficiency of the enzyme](https://image.slidesharecdn.com/2bf2eb75-f34e-4c8f-9abe-4b3fc5b9d875-161028110507/85/23-BONE-PATHOLOGIES-LAMBERT-209-320.jpg)







































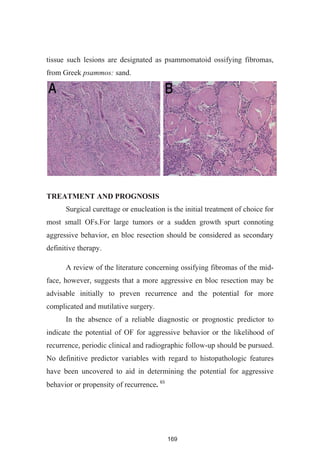
![246
MALIGNANCIES OF THE JAW93,94,95,96
PLASMA CELL NEOPLASMS
MULTIPLE MYELOMA93,96
Multiple myeloma (also known as myeloma, plasma cell
myeloma, or as Kahler's disease) is a type of cancer of plasma cells. First
described in 1848, multiple myeloma is a disease characterized by a
proliferation of malignant plasma cells and a subsequent overabundance of
monoclonal paraprotein.
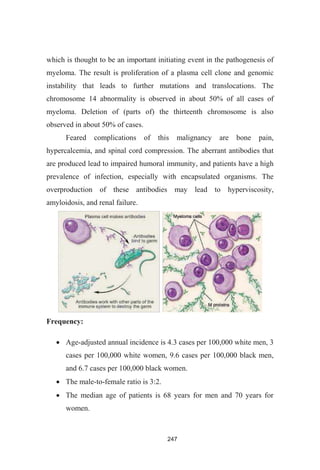
Pathophysiology
Myeloma begins when a plasma cell becomes abnormal. The
abnormal cell divides to make copies of itself. The new cells divide again
and again, making more and more abnormal cells. The abnormal plasma
cells are myeloma cells. Myeloma cells make antibodies called M proteins.
In time, myeloma cells collect in the bone marrow. They may crowd
out normal blood cells. Myeloma cells also collect in the solid part of the
bone. The disease is called "multiple myeloma" because it affects many
bones. (If myeloma cells collect in only one bone, the single mass is called
a plasmacytoma.)
The proliferation of plasma cells may interfere with the normal
production of blood cells, resulting in leukopenia, anemia, and
thrombocytopenia. The cells may cause soft tissue masses
(plasmacytomas) or lytic lesions in the skeleton. A chromosomal
translocation between the immunoglobulin heavy chain gene (on the
fourteenth chromosome, locus 14q32) and an oncogene (often 11q13,
4p16.3, 6p21, 16q23 and 20q11[6]
) is frequently observed in patients with
multiple myeloma. This mutation results in dysregulation of the oncogene](https://image.slidesharecdn.com/2bf2eb75-f34e-4c8f-9abe-4b3fc5b9d875-161028110507/85/23-BONE-PATHOLOGIES-LAMBERT-251-320.jpg)

































































































































This document provides an overview of bone pathologies and classifications of bone diseases. It begins with an introduction to bone diseases and classifications including by tissue affected, metabolic basis, and decreased/increased bone mass. It then covers specific conditions like skeletal dysplasias, metabolic bone diseases, infections, tumors, fractures and cysts. The document also provides the WHO classification of bone tumors and classifications of fibro-osseous lesions. In summary, the document classifies and describes a wide range of bone diseases, conditions, and lesions.































































