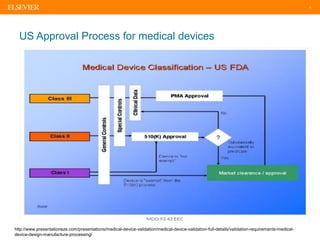
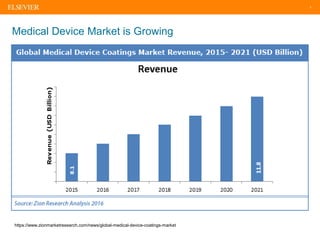
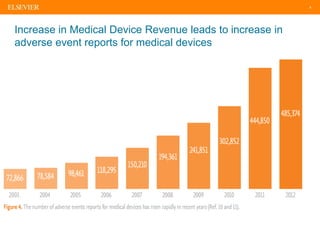
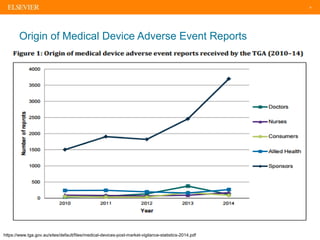
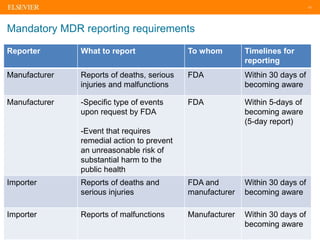
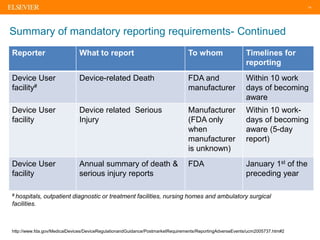
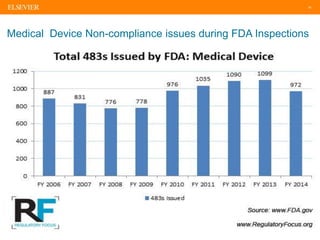
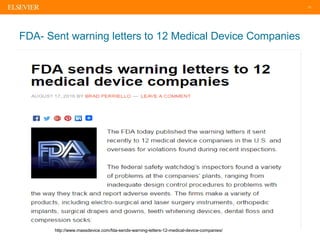
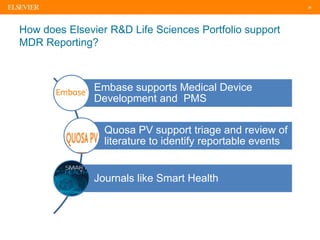
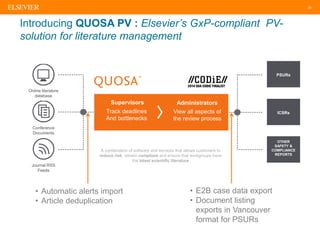
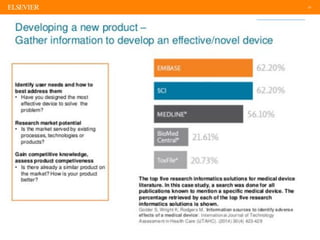
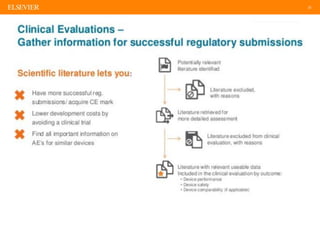
The document discusses medical device adverse event reporting requirements, including definitions of reportable events and timelines for submitting reports to regulatory agencies. It provides an overview of the classification system for medical devices and regulations around reporting malfunctions, deaths and serious injuries caused by devices. Reporting requirements and challenges involving software as a medical device are also reviewed.



































![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)









































































