Download as PDF, PPTX






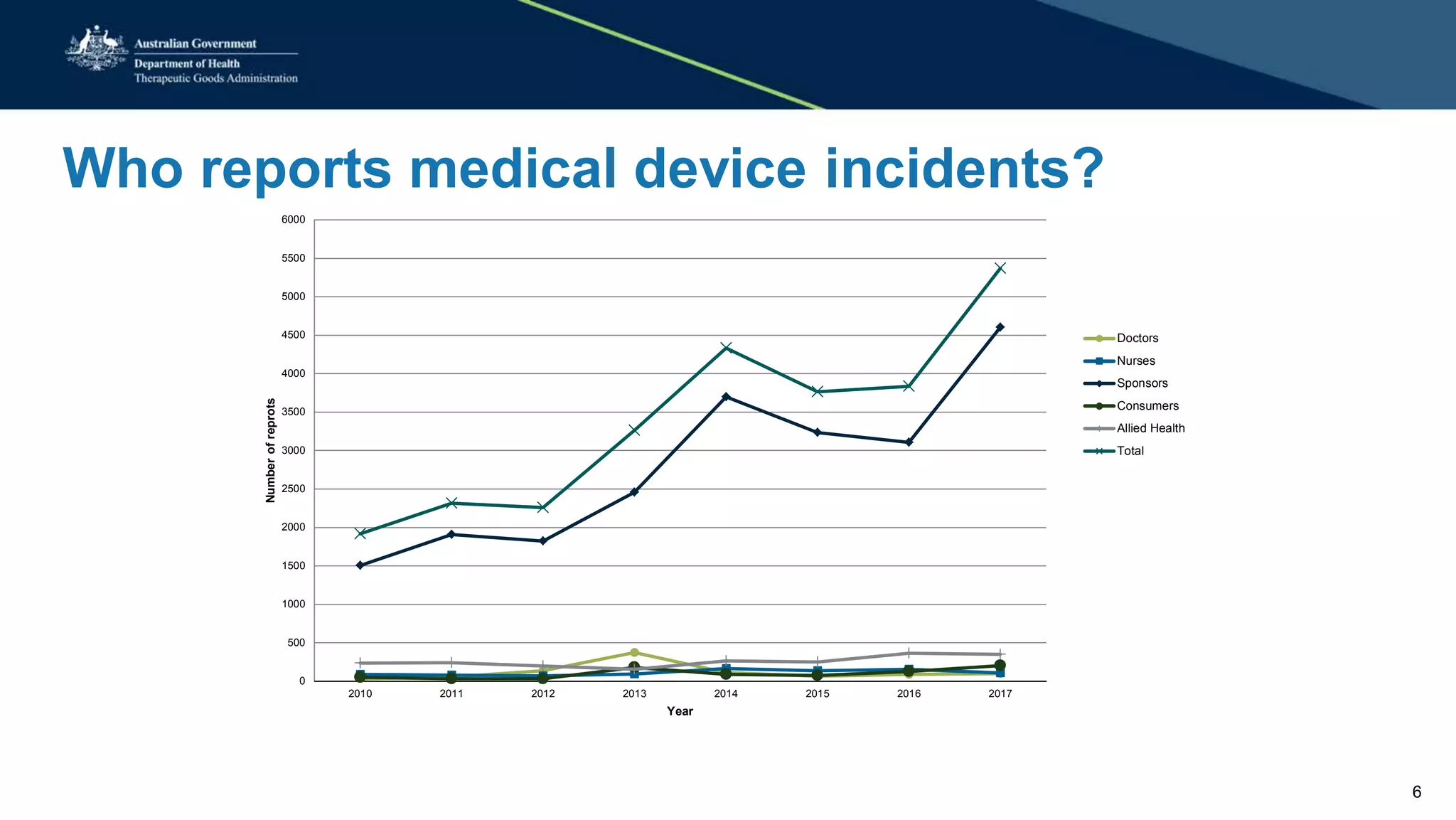


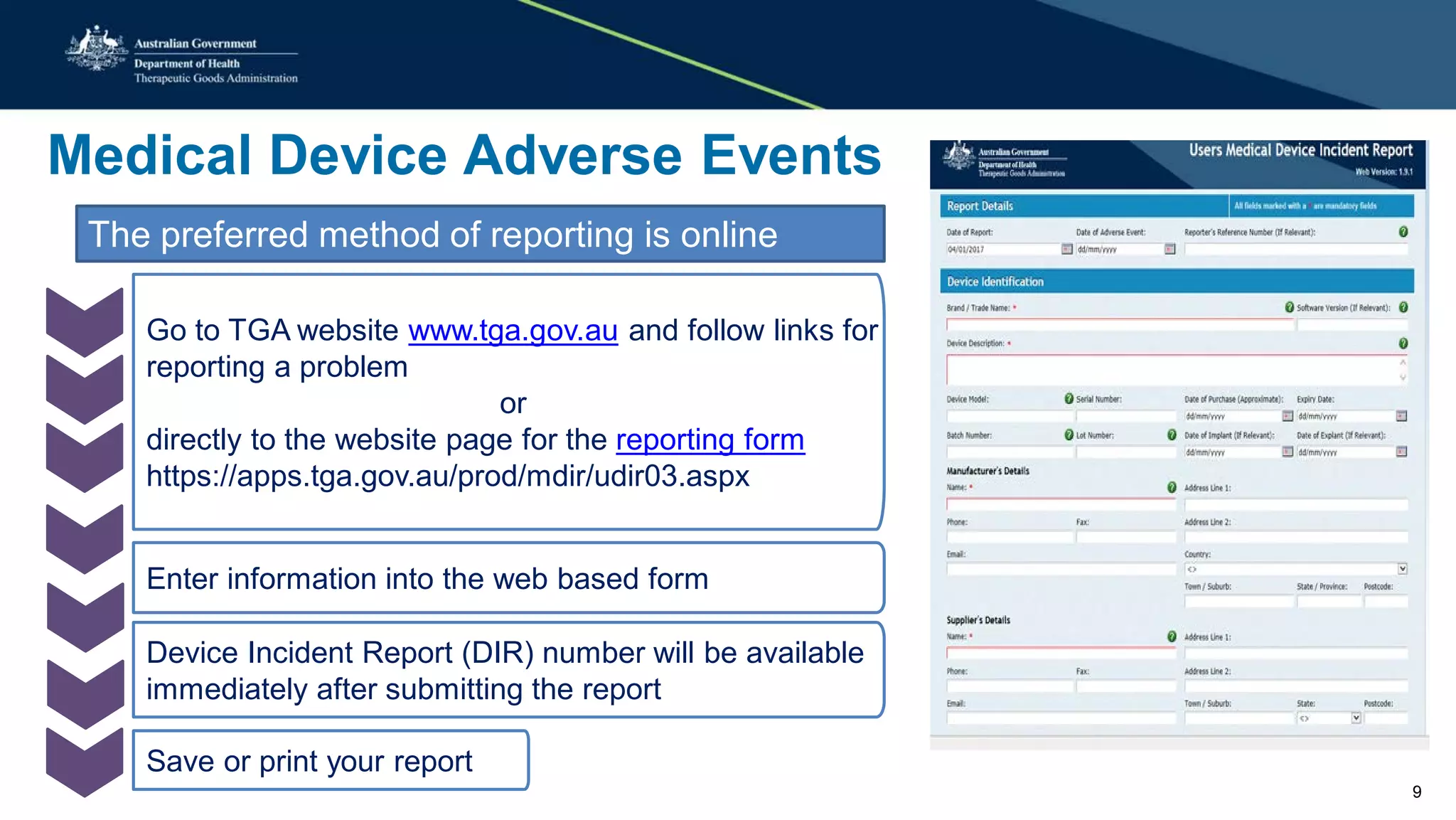




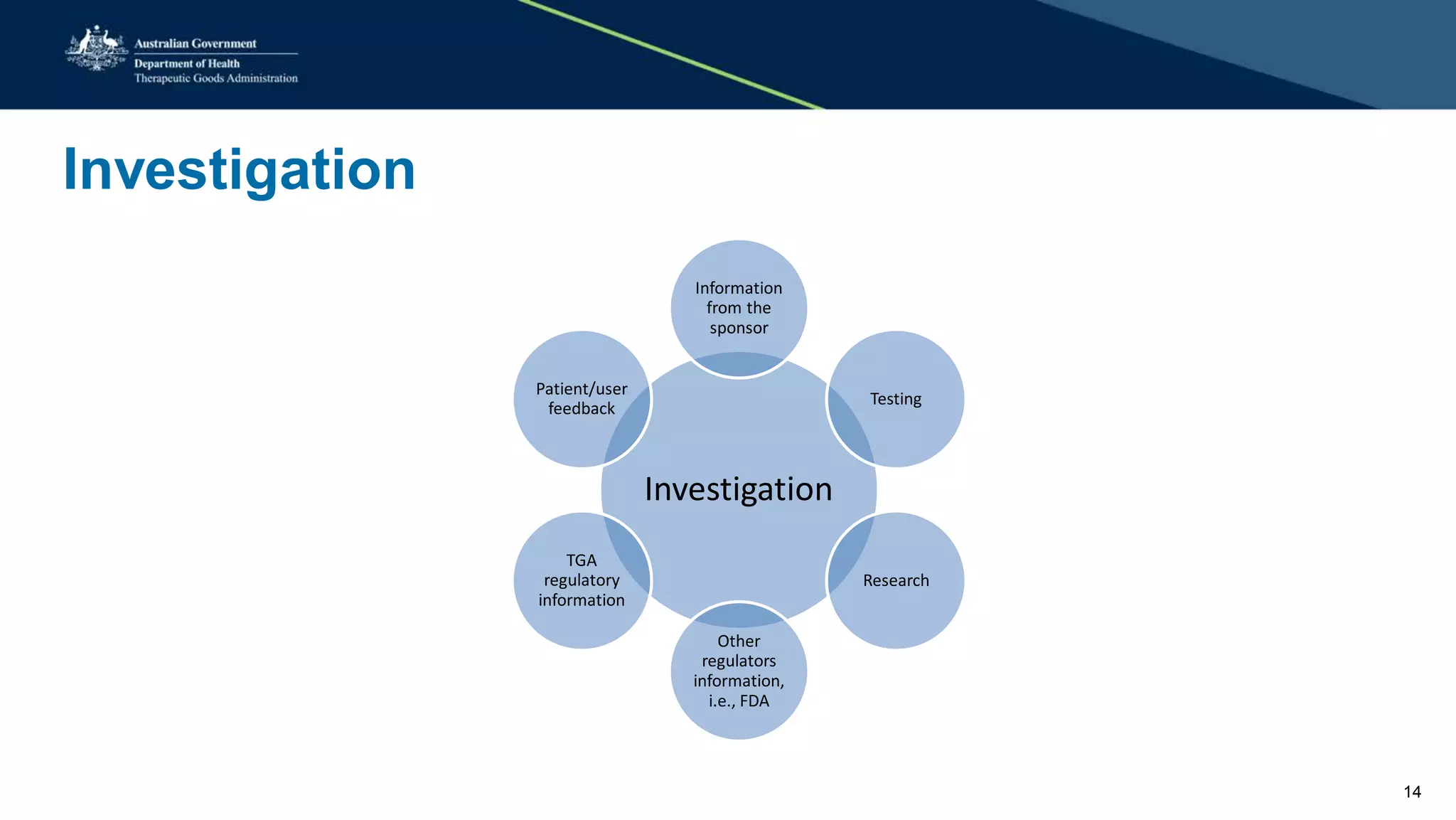








This document summarizes a workshop on reporting medical device adverse events. It outlines who is required to report events, how to submit reports, and what happens after a report is filed. The TGA evaluates reports based on severity and likelihood, and may investigate further through testing or requesting more information from manufacturers. Potential outcomes include safety alerts, recalls, or changes to device labeling. Stricter post-market monitoring of devices was also recommended to improve adverse event analysis and information sharing between regulators.





















































































