This document discusses nutrition support for various inborn errors of protein metabolism, including amino acid disorders and organic acid disorders. Key points include:
- Treatment involves restricting intake of specific amino acids or proteins to reduce toxic metabolite buildup, while providing adequate nutrition for growth. Formula supplementation provides most protein/nutrients.
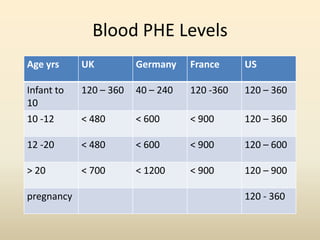
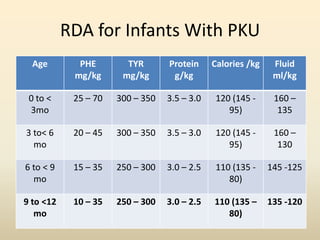
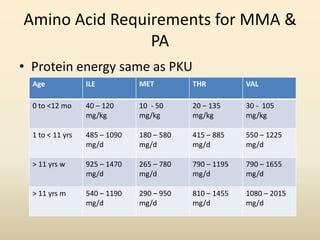
- Requirements for disorders like PKU, MSUD, and others vary by age but involve restricting intake of certain amino acids while meeting protein and calorie needs. Blood amino acid levels must be carefully monitored.
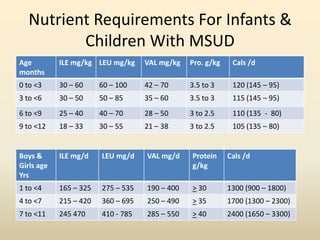
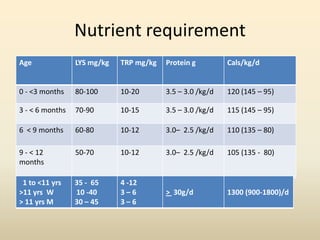
- Organic acid disorders also involve restricting intake of specific amino acids derived from lysine or tryptophan to control toxic metabolite levels while meeting nutritional needs. Early treatment is