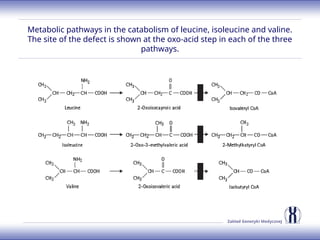
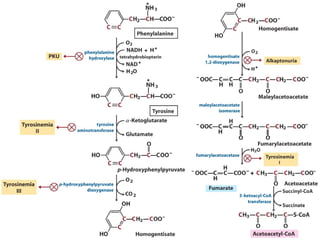
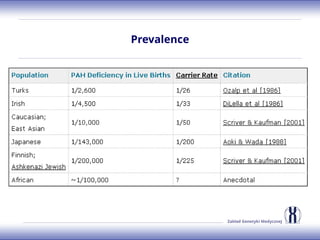
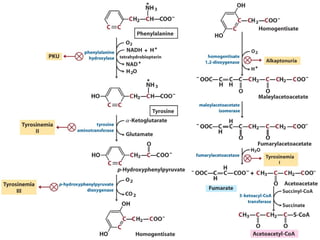
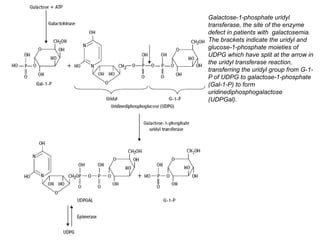
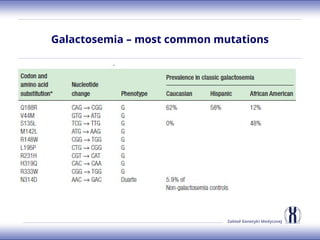
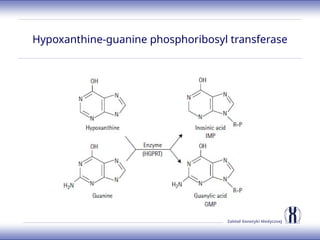
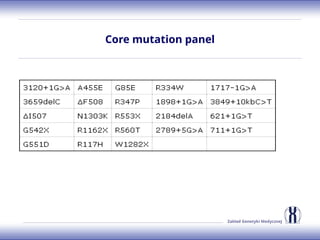
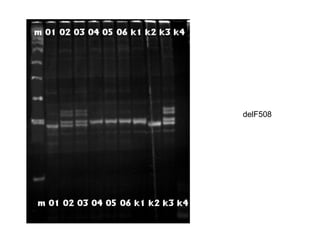
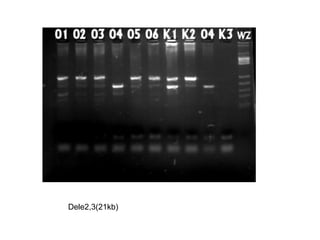
The document discusses various metabolic disorders, including organic acidemias, amino acid metabolism disorders, and fatty acid metabolism disorders, detailing their genetic basis, clinical features, prevalence, and diagnostic methods. It highlights specific conditions such as Maple Syrup Urine Disease (MSUD) and Phenylketonuria (PKU), including their symptoms, genetic mutations, and treatment options. The document also covers other disorders like galactosemia and Lesch-Nyhan disease, outlining their manifestations and the importance of dietary management.