![USFDA Approval Process
For
Drug Products & Biological Product
[NDA Vs. BLA]
By:
•
Girish A. Swami, (M.Pharm, PGDIPR, PGDDRA)
•
Mob.: +91-9881492626
•
Email- pr.girish@gmail.com](https://image.slidesharecdn.com/usfdandavsbla-140309062408-phpapp02/85/USFDA-NDA-Vs-BLA-1-320.jpg)



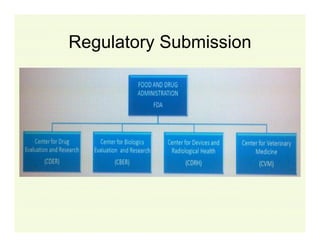




















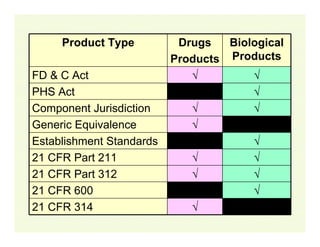






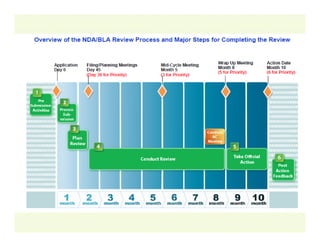








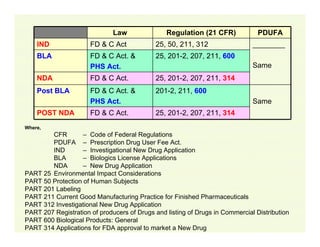




















The document provides a comprehensive overview of the USFDA approval process for drug products and biological products, detailing the roles of various centers within the FDA and the differences between New Drug Applications (NDA) and Biologics License Applications (BLA). It highlights the complexity of biologics, their regulatory requirements, and the unique challenges associated with their approval compared to traditional drugs. The document also discusses combination products, establishment standards, and post-approval requirements under the FDA framework.























































































