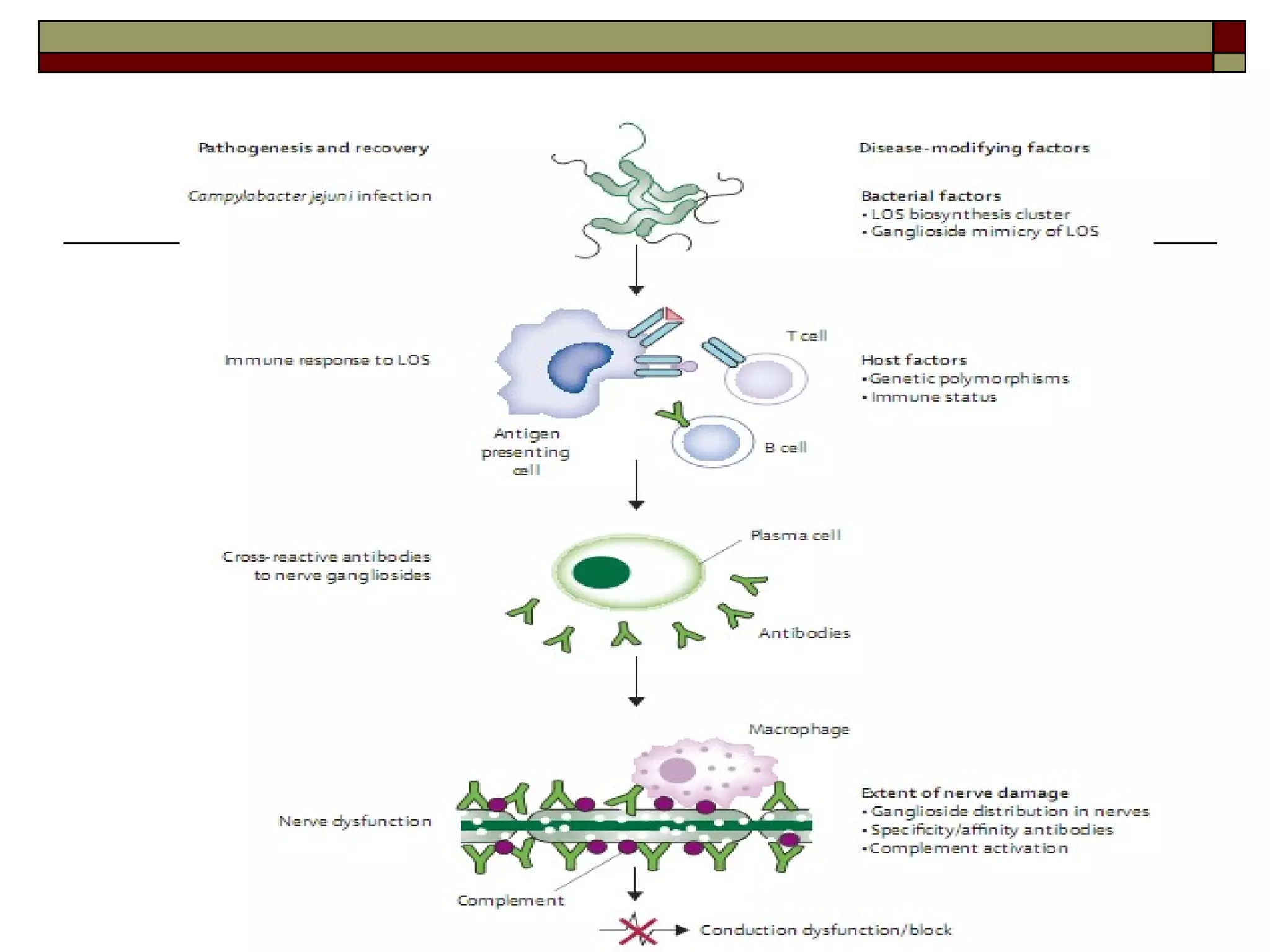
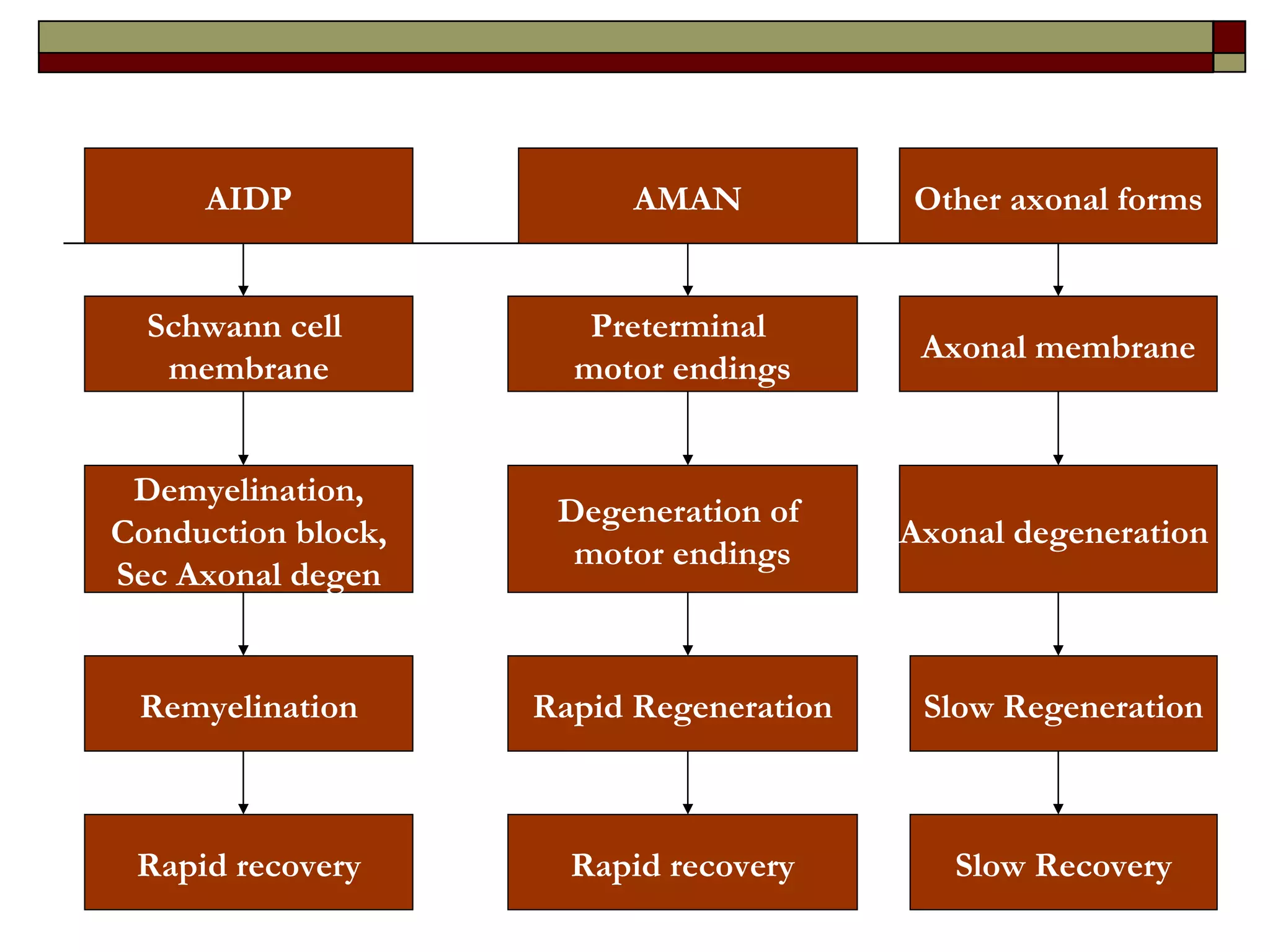
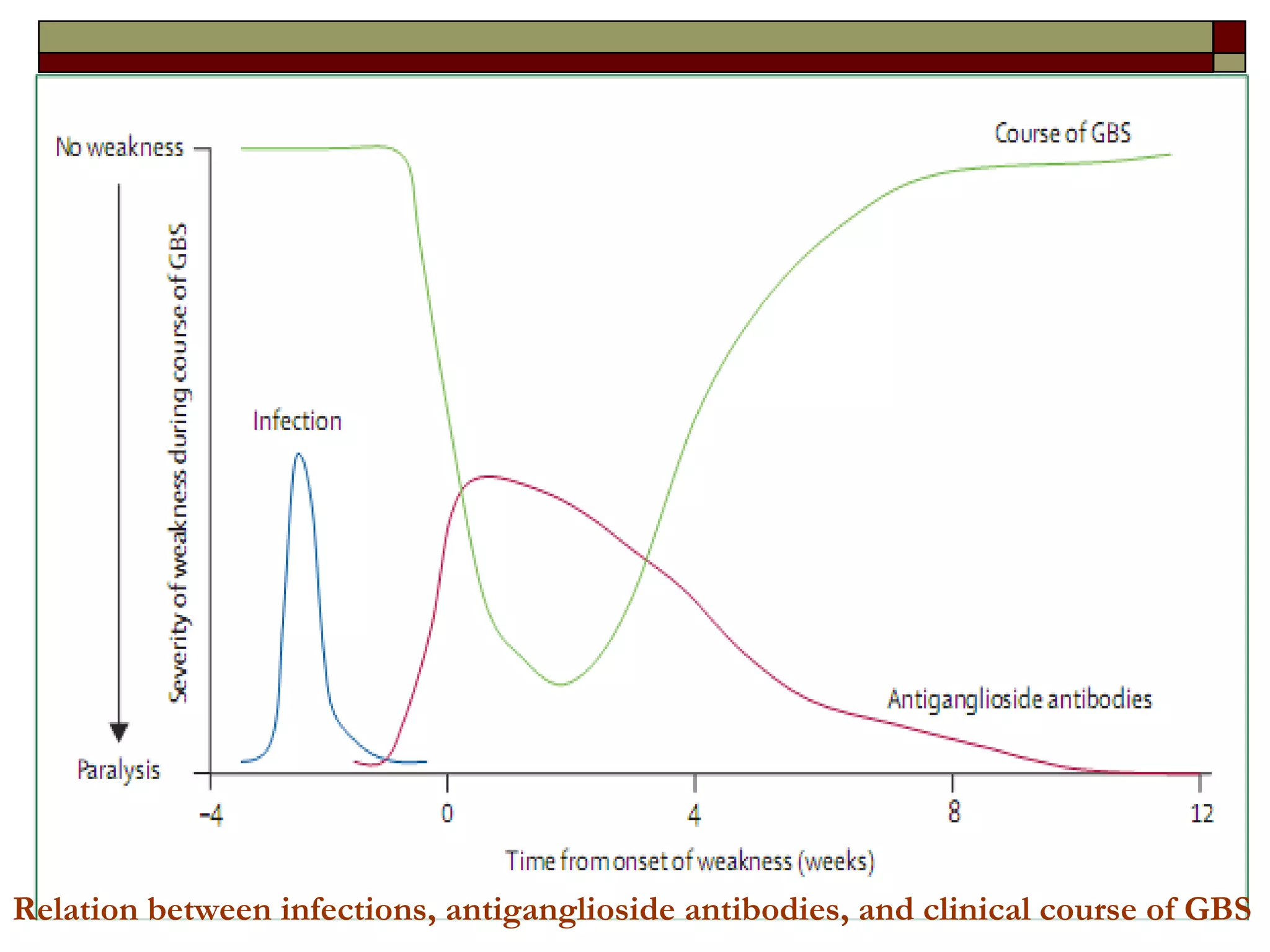
Guillain Barre Syndrome (GBS) is an acute immune-mediated inflammatory neuropathy. It is the most common cause of acute flaccid paralysis worldwide. Recent decades have seen progress in understanding the epidemiology, pathogenesis, and prognosis of GBS. The pathogenesis involves molecular mimicry between gangliosides and antigens from preceding infections like Campylobacter jejuni, leading to anti-ganglioside antibody production and complement-mediated nerve damage. Different GBS subtypes are associated with different antiganglioside antibodies and clinical courses.


































![CSF Analysis Eevated CSF protein [100–1000 mg/dL] without pleocytosis (<10 cells/mm³) is a distinctive finding. CSF is often normal in the first week; protein is elevated in 90% by the end of first week. van der Meché FG et al. Eur Neurol 2001; 45: 133–39. In a series of patients with MFS, 25% of patients had raised CSF protein in Week 1 and 84% in Week 3. Nishimoto Y et al. J Neuroimmunol 2004; 148: 200–05. CSF protein in patients with MFS and APBCN are generally lower and most often normal. No association between protein levels and outcome.](https://image.slidesharecdn.com/gbsyndrome-100223121934-phpapp01/75/Guillain-Barre-Syndrome-39-2048.jpg)


![Diagnostic Criteria for GB Syndrome ºExcluding M. Fisher and other variant syndromes Modified from AK Asbury, DR Cornblath: Ann Neurol 1990; 27: S21, 1990. 6. Electrophysiologic evidence of demyelination 3. Facial or other cranial nerve involvement 5. Typical CSF profile (cytoalbumin dissociation) 2. Mild sensory involvement 4. Absence of fever 1. Relatively symmetrical weakness Supportive 4. Exclusion of other causes [e.g., vasculitis, toxins, botulism, diphtheria, porphyria, localized spinal cord or cauda equina syndrome] 3. Disease course < 4 weeks 2. Areflexia 1. Progressive weakness of 2 or more limbs due to neuropathyº Required](https://image.slidesharecdn.com/gbsyndrome-100223121934-phpapp01/75/Guillain-Barre-Syndrome-42-2048.jpg)




































































































































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)







