Downloaded 20 times

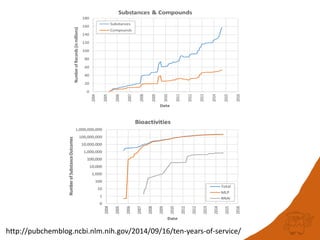
![Big Data is…
“…a broad term for data sets so large or complex
that traditional data processing applications are
inadequate.” [Wikipedia]
Any dataset could be considered Big Data
without sufficiently efficient algorithms and
tools](https://image.slidesharecdn.com/rscapr2015bigdata-150423041734-conversion-gate02/85/Is-20TB-really-Big-Data-2-320.jpg)


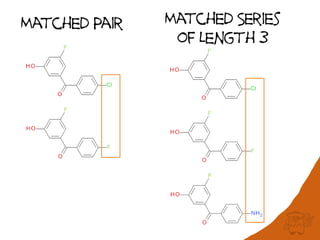



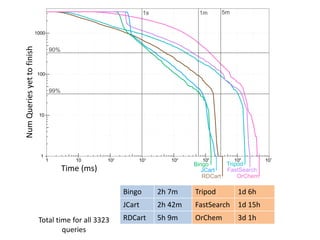
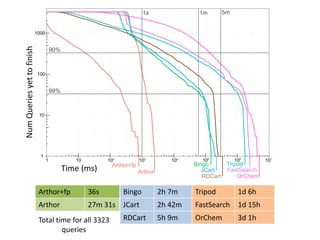






![Performance
• Scales with the number of SMARTS patterns
used
– currently 802 patterns for 504 reactions
• Test set: 1.1 million reactions extracted from
the USPTO applications 2001-2012
– Processed in 11.2 h
– 437 K reactions named
US20010000038A1 : 3.10.2 Friedel-Crafts alkylation [Friedel-
Crafts reaction]
US20010000038A1 : 10.1.5 Wohl-Ziegler bromination
[Halogenation]](https://image.slidesharecdn.com/rscapr2015bigdata-150423041734-conversion-gate02/85/Is-20TB-really-Big-Data-21-320.jpg)
[C@@H](C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc2c[nH]cn2)C(=
O)N3CCC[C@H]3C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(=O)O)C(=O)N4CCC[C@H]4C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(
C)C)C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](C)C(=O)N[C@@H](CS)C(=O)N[
C@@H](CCC(=O)O)C(=O)N5CCC[C@H]5C(=O)N6CCC[C@H]6C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](CC(=O)N)C(=O)N[C@
@H](C(C)C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(=O)
O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CO)C(=
O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc7c[nH]c8c7cccc8)C(
=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CCC(=O)N)C(=O)N9CCC[C@H]9C(=O)N[C@@H](C)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[
C@@H](CC(=O)O)C(=O)NCC(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](
[C@@H](C)O)C(=O)NCC(=O)N[C@@H](Cc1ccc(cc1)O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC
(=O)O)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC(C)C)C(=O)N1C
CC[C@H]1C(=O)N[C@@H](CC(=O)O)C(=O)NCC(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](Cc1c[nH]c2c1cccc2)C(=O)N[C@@H](
[C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H]([C@@H
](C)O)C(=O)N[C@@H](CC(=O)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@
H]([C@@H](C)O)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(
=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](CC
(=O)N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(=O)N)C(=O)N[C@@H](Cc1ccc(cc1)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H
](Cc1ccccc1)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](C)C(=O)……](https://image.slidesharecdn.com/rscapr2015bigdata-150423041734-conversion-gate02/85/Is-20TB-really-Big-Data-22-320.jpg)




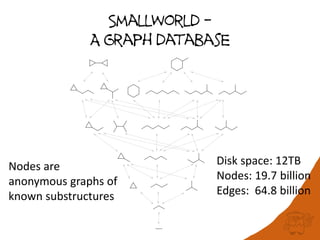
100 million compounds, 100K protein structures, 2 million reactions, 1 million journal articles, 20 million patents and 15 billion substructures. Is 20TB really Big Data? With modern hardware and efficient algorithms, many classic cheminformatics problems can be handled with today’s datasets. Noel O’Boyle, Daniel Lowe, John May and Roger Sayle of NextMove Software discuss how traditional cheminformatics tasks can be performed on large chemical datasets through techniques like precomputing substructures, optimised substructure searching, and graph databases.





























![RDKit: Six Not-So-Easy Pieces [RDKit UGM 2016]](https://cdn.slidesharecdn.com/ss_thumbnails/rdkittautomersbasel16-161031122400-thumbnail.jpg?width=640&height=640&fit=bounds)





































































