Download as PDF, PPTX










(Cl)(F)Br should be the same as
[C@@](O)(Cl)(F)1.Br1 (if supported)](https://image.slidesharecdn.com/oboyle-smilesbenchmark-180819144958/85/A-de-facto-standard-or-a-free-for-all-A-benchmark-for-reading-SMILES-10-320.jpg)










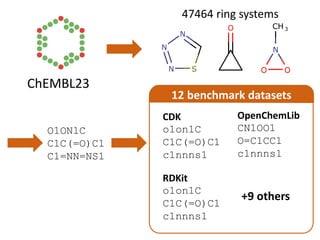

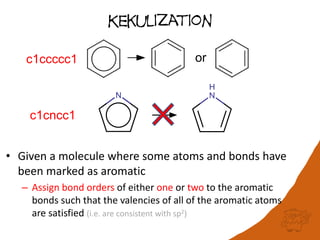
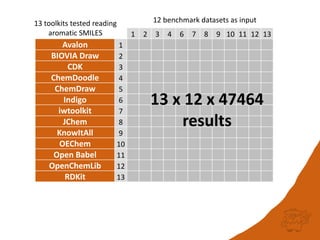
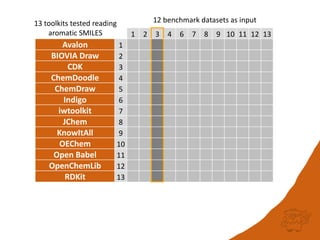
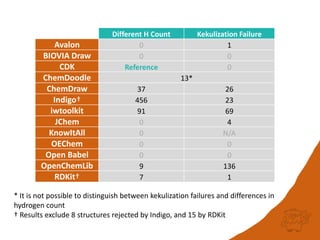
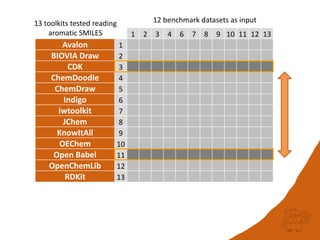








The document discusses a benchmark for evaluating how accurately different cheminformatics toolkits can read SMILES strings representing stereochemistry, implicit hydrogens, and aromatic systems. The author tested 15 toolkits on test cases examining cis-trans stereochemistry, implicit valence as defined by the SMILES specification, and their ability to consistently interpret the aromatic nature and hydrogen counts of molecules represented by SMILES strings. While stereochemistry is generally handled well, adherence to the SMILES valence model and consistent aromatic interpretation vary more between tools. The benchmark aims to identify such differences and facilitate improvements to interoperability.













































![RDKit: Six Not-So-Easy Pieces [RDKit UGM 2016]](https://cdn.slidesharecdn.com/ss_thumbnails/rdkittautomersbasel16-161031122400-thumbnail.jpg?width=640&height=640&fit=bounds)



















![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)

