
Recommended
More Related Content
What's hot
What's hot (20)
Similar to JBEI Research Highlights - November 2018
Similar to JBEI Research Highlights - November 2018 (20)
More from Irina Silva
More from Irina Silva (19)
Recently uploaded
Recently uploaded (20)
JBEI Research Highlights - November 2018
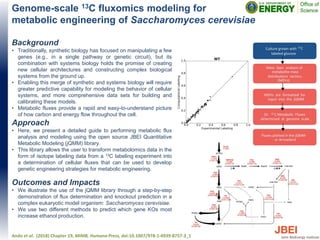
- 1. Genome-scale 13C fluxomics modeling for metabolic engineering of Saccharomyces cerevisiae Background • Traditionally, synthetic biology has focused on manipulating a few genes (e.g., in a single pathway or genetic circuit), but its combination with systems biology holds the promise of creating new cellular architectures and constructing complex biological systems from the ground up. • Enabling this merge of synthetic and systems biology will require greater predictive capability for modeling the behavior of cellular systems, and more comprehensive data sets for building and calibrating these models. • Metabolic fluxes provide a rapid and easy-to-understand picture of how carbon and energy flow throughout the cell. Approach • Here, we present a detailed guide to performing metabolic flux analysis and modeling using the open source JBEI Quantitative Metabolic Modeling (jQMM) library. • This library allows the user to transform metabolomics data in the form of isotope labeling data from a 13C labeling experiment into a determination of cellular fluxes that can be used to develop genetic engineering strategies for metabolic engineering. Outcomes and Impacts • We illustrate the use of the jQMM library through a step-by-step demonstration of flux determination and knockout prediction in a complex eukaryotic model organism: Saccharomyces cerevisiae. • We use two different methods to predict which gene KOs most increase ethanol production. Ando et al. (2018) Chapter 19, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_1
- 2. Microbial metabolomics: a general overview Background • In the biosciences, there is growing interest elucidating gene function. Consequently, metabolomics has garnered a lot of attention due to its provision of metabolic information pertaining to both function and phenotype. • This chapter briefly introduces a few important aspects of the metabolome, the challenges faced when capturing metabolomic information and the steps that are necessary to overcoming them. Approach • Metabolomics seeks to measure the low molecular weight chemical products of enzyme-catalyzed reactions (metabolites) in biological systems (e.g., cells, compartments, tissues, organisms) at specific points in time. Outcomes and Impacts • Metabolites are distinct chemical entities from biochemical transformations, which complicates both the measurement and data analysis processes. As a result, there is no single sample preparation or analytical method that can extract or measure an entire microbial metabolome. • While nuclear magnetic resonance provides high throughput metabolic fingerprinting and unambiguous metabolite identification, mass spectrometry is more popular as it provides quantitative information at reduced costs. • At present, metabolomics data is more widely applied to human related studies, followed by plant studies, and then microbiological studies. • Metabolomics is becoming a key component of systems and synthetic biology studies, and its application has been extended to microbial community research. • When metabolomics is part of an integrated multi-omics approach the data generated can be used to produce accurate predictive models for systems microbiology and synthetic biology research. Baidoo et al. (2018) Chapter 1, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_1
- 3. Mass spectrometry-based microbial metabolomics: techniques, analysis, and applications Background • The demand for understanding the roles genes play in biological systems has steered the biosciences into the direction the metabolome, as it closely reflects the metabolic activities within a cell. • Mass spectrometry (MS) based metabolomic information is being used to characterize microbial metabolic networks and has made significant contributions to microbiological research in the environment and human disease. • In this chapter, the technical aspects of metabolomics are discussed and as well as its application to microbiological research. Approach • Following the quenching of microbial metabolism, metabolites are extracted (e.g. via liquid-liquid, SPE, SPME, etc.) and preserved for MS data acquisition and analysis. Outcomes and Impacts • The chemical diversity within the metabolome has led to the adoption of targeted metabolite extraction approaches by most laboratories rather than a universally applied method. • The acquisition of high resolution accurate MS (e.g. TOF and orbitrap) data provides qualitative and quantitative information, with a targeted metabolomics approach being favored by most research laboratories. • Quantitative metabolomic information is easily obtained when chromatographic and electrophoretic separation is coupled to MS. • Metabolomics techniques are applied to metabolic flux analyses, systems and synthetic biology, environmental microbiology (e.g. microbial community studies), and human disease. • While the scientific community is willing to put significant resources behind ‘omics’ approaches, the quality of metabolomics data going forward will determine the role it will play in future microbiological research. Baidoo et al. (2019) Chapter 2, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_2
- 4. Probabilistic lifecycle assessment of butanol production from corn stover using different pretreatment methods Background • Studies on cellulosic butanol have only considered sulfuric acid pretreatment process and many of these studies present deterministic results. • This study seeks to bridge the research and modeling gaps by developing stochastic process model integrating feedstock supply logistics and the downstream butanol production process, and considering the five most commonly considered biomass deconstruction methods (e.g., steam explosion, sulfuric acid, ammonia fiber explosion [AFEX], ionic liquid [IL] and biological) Approach • We developed stochastic lifecycle assessment models and determined lifecycle energy use and greenhouse gas emissions. • We demonstrated performance targets for future research. Outcomes and Impacts • Probabilistic results of these analyses describe a distribution of GHG emissions with an average of 18.09-1056.12 gCO2e/MJ and a 95% certainty to be less than 33.3-1888.3 gCO2e /MJ. • The highest GHG emissions of IL-pretreatment of 1056.12 gCO2e/MJ reaches to 89.8 gCO2e/MJ by switching IL-recovery from 80 to 99 wt%, which is the most influencing parameter for IL-pretreatment. • We propose alternative ionic liquid (IL) including cholinium lysinate and triethylammonium hydrogen sulfate, as these ILs could reduce the carbon footprint of IL-based biomass deconstruction process. • When the influential inputs can be optimized, many of these pretreatment methods can be used to realize GHG emissions and net energy reduction goals. Baral et al. (2018) Environ. Sci. Technol., doi: 10.1021/acs.est.8b05176
- 5. Model metabolic strategy for heterotrophic bacteria in the cold ocean based on Colwellia psychrerythraea 34H Background • Colwellia psychrerythraea 34H is a model psychrophilic extremophile bacterium (unable to grow above 18 C) found in the cold ocean—polar sediments, sea ice, and the deep sea. • Genomic studies of 34H and other strains of C. psychrerythraea have revealed many metabolic pathways that are important in the geochemical cycling of nutrients in cold marine environments (e.g. degradation of components of natural gas). • Although the genomes of such psychrophiles have been sequenced, their metabolic strategies at low temperature have not been quantified. Approach • We measured the metabolic fluxes (using 13C-fingerprinting and 13C- MFA) and gene expression of 34H at 4 C (the mean global-ocean temperature and a normal-growth temperature for 34H), making comparative analyses at room temperature (above its upper-growth temperature of 18 C) and with mesophilic Escherichia coli. Outcomes and Impacts • When grown at 4 C, 34H utilized multiple carbon substrates without catabolite repression or overflow byproducts; its anaplerotic pathways increased flux network flexibility and enabled CO2 fixation. • In glucose-only medium, the Entner–Doudoroff (ED) pathway was the primary glycolytic route; in lactate-only medium, gluconeogenesis and the glyoxylate shunt became active. • Consequently, ED pathway expression levels were much higher than the upper portion of the (Embden–Meyerhof–Parnas) EMP pathway. • Specific findings have relevance to bioremediation of pollutants from the petroleum industry and to biomanufacturing of cold-adapted enzymes. Czajka et al. (2018) PNAS, doi: 10.1073/pnas.1807804115 Metabolic flux map when 34H was grown at 4 C on 1,2-13C glucose (Left) or 3-13C lactate (Right). Transcriptomic expression data of 34H at normal-growth conditions and differential expression data after exposure to temperature-stressed conditions as determined by RNA- Seq.
- 6. Cluster analysis of untargeted metabolomic experiments Background • Untargeted metabolite profile data based upon liquid chromatography-based mass spectrometry (LC-MS) represents dense biological data. • Cluster analysis, or clustering, is a common technique in data mining and used in many fields such as machine learning, data compression and computer graphics. • Here we examine the use of Principal Component Analysis and Hierarchical Clustering, two common cluster analysis tools, for digging into multi-dimensional metabolite data. Approach • Dense data refers to each replicate having hundreds to thousands of different variables or molecular feature metabolites, which are represented quantitatively. • Orthogonal transformation is a linear transformation which preserves a symmetric inner product. In general, an orthogonal transformation preserves the lengths of vectors and angles between vectors. Outcomes and Impacts • PCA can be used to map metabolites by their similarity, however we are using them to group replicates in our example. The former results in more of a cloud as there are thousands of metabolites in the data files. • Unsupervised analysis is much better for getting an objective view of your data as you do not feed any class information to the algorithm. • Different values can be given for plot3D variables to allow you to customize the plot how you like visually. J. Heinemann (2018) Chapter 16, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_11 PCA plots of first three principal components from ten biological replicates of S. solfataricus before and after H2O2 stress, three variants including (a) media 1 before and after 30 min 100 mM H2O2, (b) Media 1 before and after 30 min 30 μM H2O2, (c) media 2 before and after 30 min 30 μM H2O2, and (d) all experiments combined
- 7. Machine learning in untargeted metabolomics experiments Background • Machine learning (ML) is a form of artificial intelligence (AI) that provides computers with the ability to learn generally without being explicitly programmed. • Outputs of machine learning algorithms can be grouped into classification, regression, clustering, and dimensionality reduction. • When analyzing untargeted metabolomics data with ML, there is substantial risk of overfitting because the number of variables (metabolites) is greater than the number of instances (replicates), increasing the likelihood that the ML algorithm can only predict accurately within those data sets. Approach • Here we examine the use of machine learning for use with untargeted metabolomics data, when it is appropriate to use, and questions it can answer. • We provide an example workflow for training and testing a simple binary classifier, a multiclass classifier and a support vector machine using the Waikato Environment for Knowledge Analysis (Weka), a toolkit for machine learning. Outcomes and Impacts • Cross validation is only possible for supervised learning as the data categories must be known for the algorithm to run recursively on all possible splits of the data training versus testing. • Utilizing the sensitivity of metabolite abundance to stress, disease and environmental variation makes them the perfect medium for creating dense biological data suitable for analysis and further prediction using ML algorithms and workflows. J. Heinemann (2018) Chapter 17, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_11
- 8. Metabolomics: a microbial physiology and metabolism perspective Background • Metabolism is a complex sequence of reactions that direct biological functions in living things, and metabolomics enables simultaneous analysis of metabolic reactions and pathways involved in these processes. • Unlike genomics, transcriptomics and proteomics that rely on a finite set of metabolites or subunits, metabolomics evaluates the total set of metabolites in an organism. Approach • This review highlights philosophical and scientific considerations for executing effective metabolic studies. • The importance of growth in metabolic studies is often understated, but it is the primary cause of most inconsistencies in these studies. Outcomes and Impacts • Physiological studies and metabolic prediction require effective strategy for quantitative metabolite analysis and the accuracy of metabolomics makes it an ideal analytical tool for these studies. • Metabolomics provides a snapshot of metabolism at any given moment in an organism, which is further enhanced by the addition of data from other analytical strategies such as transcriptomics, proteomics, and biochemical analyses. • In addition to reproducible analytical tools, metabolic prediction requires fundamental understanding of metabolism and physiology, which is critical for experimental design and data interpretation. CJ Joshua (2018) Chapter 3, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_1 Central carbon metabolism highlighting classical glycolytic pathways, tricarboxylic acid pathway, gluconeogenesis (blue lines), glyoxylate shunt (green lines), glycerol-3-phosphate and pyruvate–lactate redox shuttle pathways (gray boxes).
- 9. Recent advances in x-ray hydroxyl radical footprinting at the Advanced Light Source synchrotron Outcomes and Impacts • Figure 1 compares the dose response curves and total ion chromatograph mass spectrometry results from a focused vs. an unfocused white-light bend magnet source at the ALS. More damage was observed for the N-terminal portion of the protein using 50 msec exposure as opposed to 0.6 msecs. • Initial “drop-on-demand” experiments were conducted using a Canon XS40HS camera to manually analyze color in 500 µm droplets falling through a ~4mm wide visible light beam (Figure 2). The change in color due to decreasing concentration of Alex488 dye, is correlated to increasing damage from an X-ray beam. • An advantage of the “drop-on-demand” and real time color monitoring technology is that it only requires 10-20 µl of sample per dose response point. • Preliminary “drop-on-demand” experiments have demonstrated that high flux density combined with container-less sample presentation to the X-ray beam enable high-dose HRF experiments, which in turn yield high quality data. Morton et al. (2018) Protein Pept. Lett., doi: 10.2174/0929866526666181128125725 Background • Efforts in the field of synchrotron hydroxyl radical footprinting (HRF) are progressing rapidly to investigate structural features and conformational changes of nucleic acids and proteins in the solution state. • Minimizing sample volume is imperative for studies in which multiple samples and many conditions must be tested. However, as the sample volume decreases, the relative X-ray dose decreases. Approach • Here we compare the dose response curves and mass spectrometry results from a focused vs. an unfocused white-light bend magnet source at the ALS. • We are also developing “drop-on-demand” methodologies to increase the dose received by the sample while maintaining microsecond exposure times. Figure 1: (Top) Dose response curves for a 40 kDa protein at a focused (left) vs unfocused (right) white-light bend magnet beamline source at the ALS. Top: comparison of exposure times necessary for the same amount of modification. Solid lines (red and black) represent pseudo unimolecular fit using full and first 3 data points respectively. Solid blue line represents a fit to y=y0+Aexp(-kt). (Bottom) Total ion chromatogram (TIC) achieved for 0 and maximum dose. More damage was observed for the N-terminal portion of the protein using 50 msec exposure as opposed to 0.6 msecs. Figure 2: High-speed camera images of falling drops with various concentrations of Alexa488 dye (left) and manual colorimetry measurements of the drops (right).
- 10. Accelerating the deployment of anaerobic digestion to meet zero waste goals Background • Feedstock cost is a significant barrier to the commercialization of bioenergy technologies. • Mixed municipal organic wastes are challenging to convert, but also offer the advantage of negative feedstock costs (tipping fees). Approach • This feature article explores the key technical and regulatory barriers to increasing the utilization of mixed municipal organics, and recommends research directions to address these challenges. • The article draws from broad experiences across the industry, literature, and detailed data and operational experiences at a dry anaerobic digestion facility in San Jose, CA (Zero Waste Energy Development). Outcomes and Impacts • Key challenges identified and explored include: (1) Sorting technologies and practices to minimize feedstock contamination, (2) Predicting biogas yields and composition from mixed waste streams, (3) Mitigating air pollution to comply with local regulations and avoid odor complaints, (4) Maximizing the economic value of energy outputs, (5) Maximizing the net climate benefits of anaerobic digestion and coproducts • Many of these challenges are broadly applicable to bioenergy systems beyond anaerobic digestion Satchwell et al. (2018) Environ. Sci. Technol., doi: 10.1021/acs.est.8b04481)
- 11. Liquid chromatography and mass spectrometry analysis of isoprenoid intermediates in Escherichia coli Background • Isoprenoids are a highly diverse group of natural products with broad application as high value chemicals and advanced biofuels. • However, the measurement of isoprenoid intermediates via standard liquid chromatography-mass spectrometry (LC-MS) protocols is challenging due to their hydrophilicity and complex physicochemical properties. • Here we describe a robust hydrophilic interaction liquid chromatography time-of-flight mass spectrometry (HILIC-TOF-MS) method for analyzing isoprenoid intermediates from metabolically engineered Escherichia coli strains. Approach • HILIC separation was mediated by hydrogen bonding between the analyte and LC stationary phase. • HILIC separation was coupled to TOF-MS via electrospray ionization and analytes were detected via high resolution accurate mass measurements of deprotonated ion adducts. • Following metabolite extraction from Escherichia coli biomass, isoprenoid intermediates were quantified by HILIC-TOF-MS. Outcomes and Impacts • The HILIC-TOF-MS method was able to resolve all but two of the isoprenoid intermediates tested (i.e., IPP/DMAPP). • The method was used to identify pathway bottlenecks in engineered Escherichia coli by observing the accumulation of intermediate metabolites (which may have occurred due to reduced activity of the proceeding enzyme(s)). • The method can also be used to monitor dephosphorylated pathway intermediates (e.g., IP/DMAP and FP), sugars, nucleotide cofactors, and central carbon metabolites of the glycolysis pathway and the tricarboxylic acid (TCA) cycle. Baidoo et al. (2018) Chapter 11, MIMB, Humana Press, doi:10.1007/978-1-4939-8757-3_11