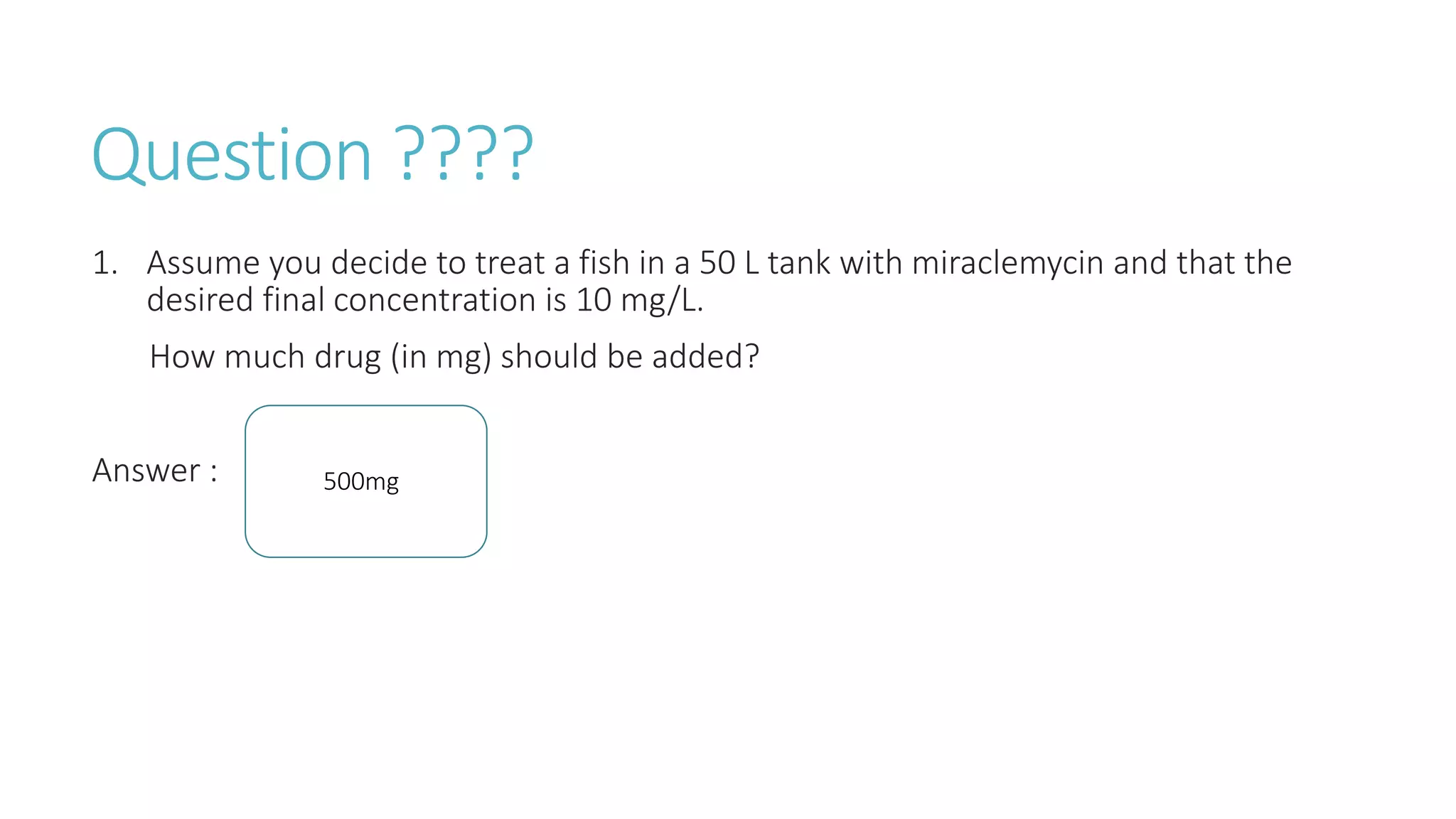
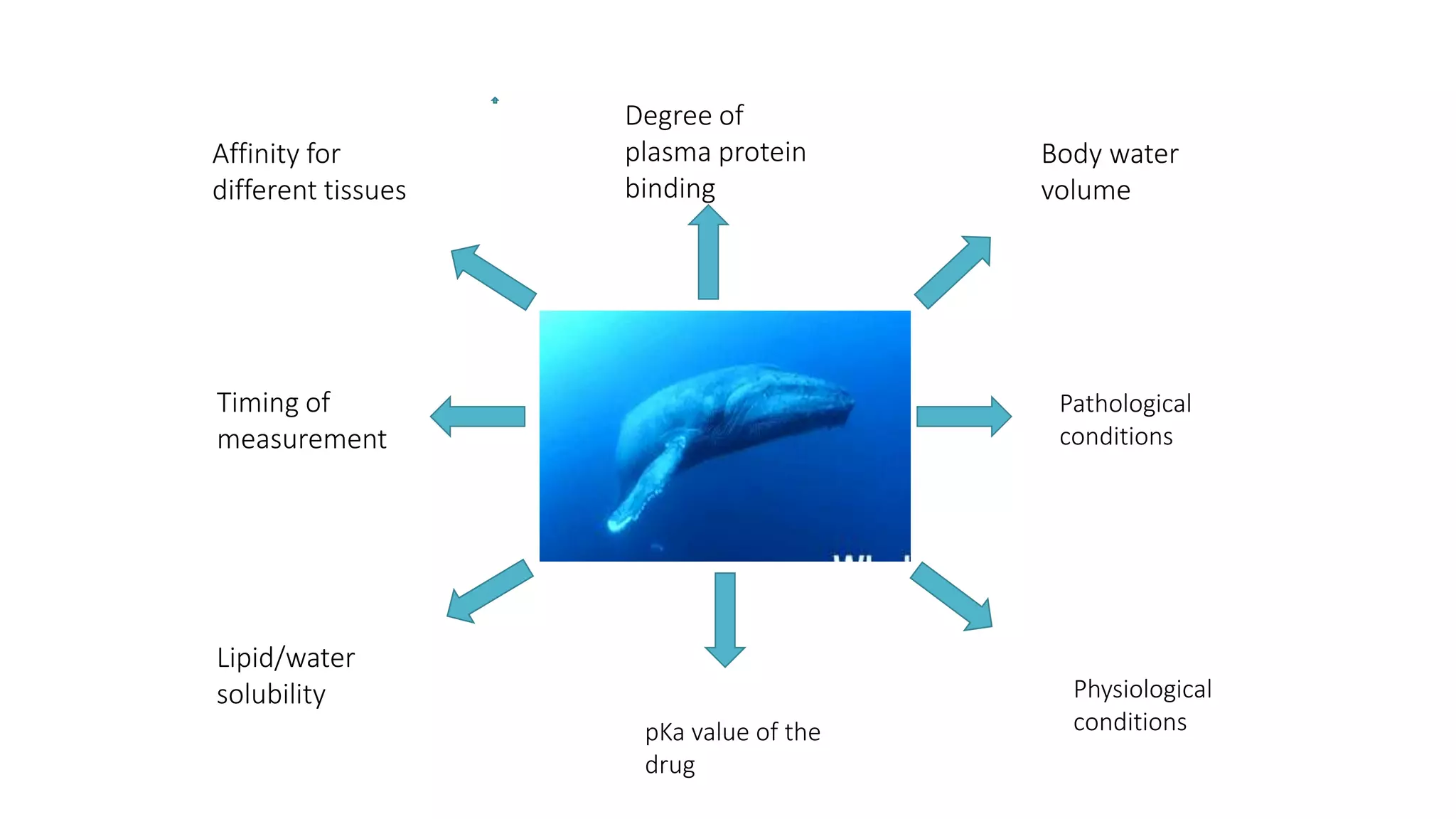
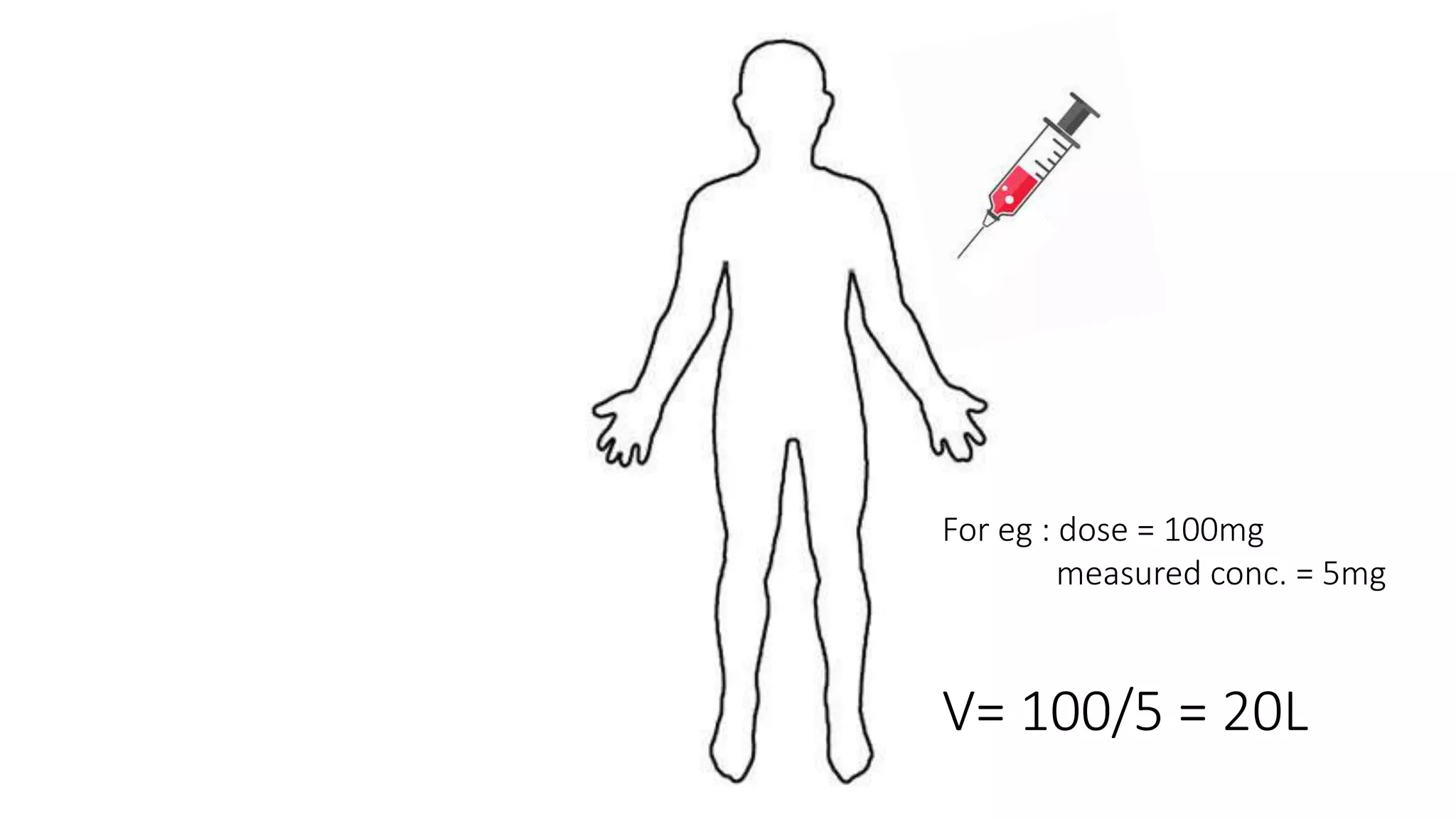
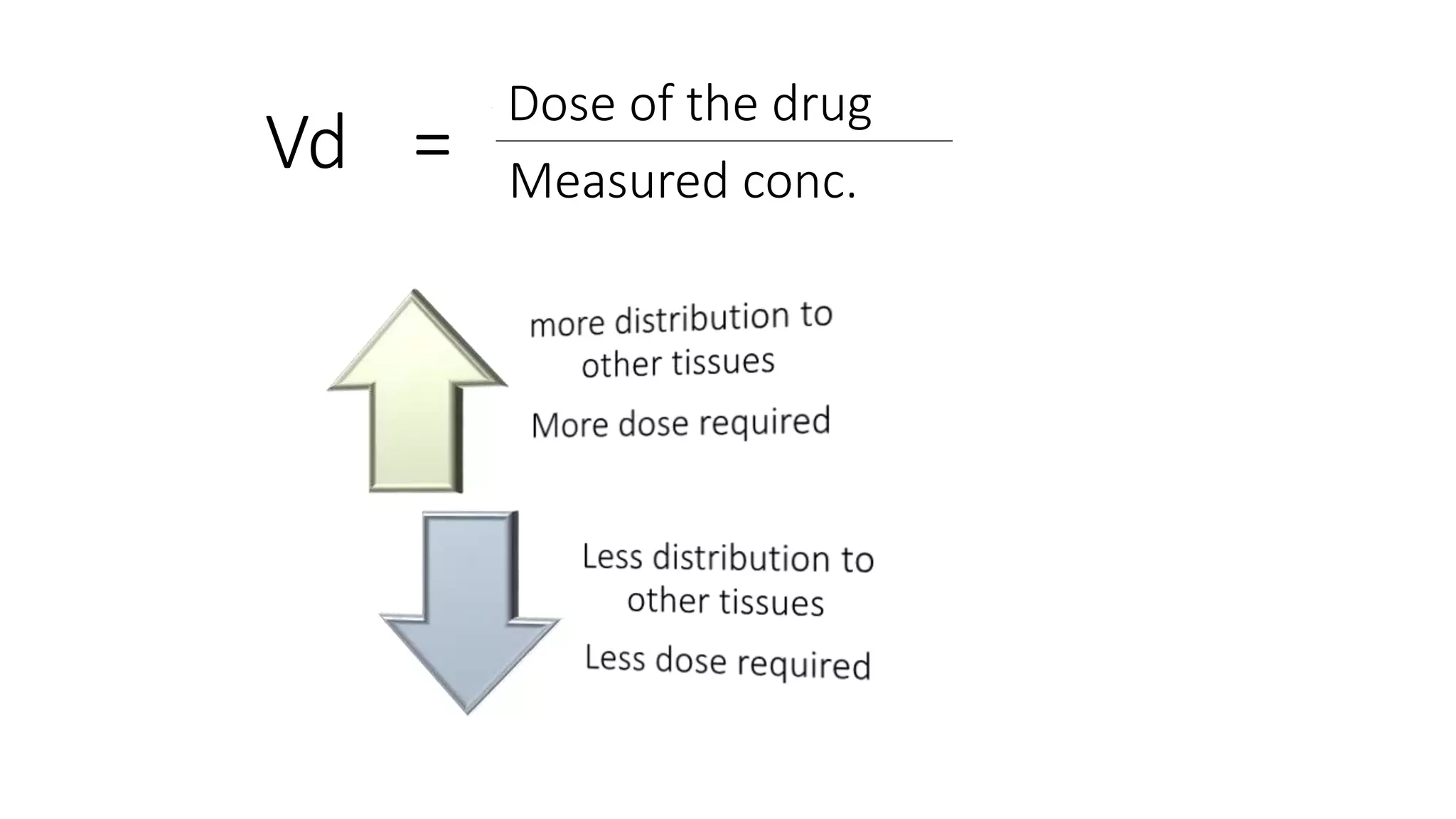
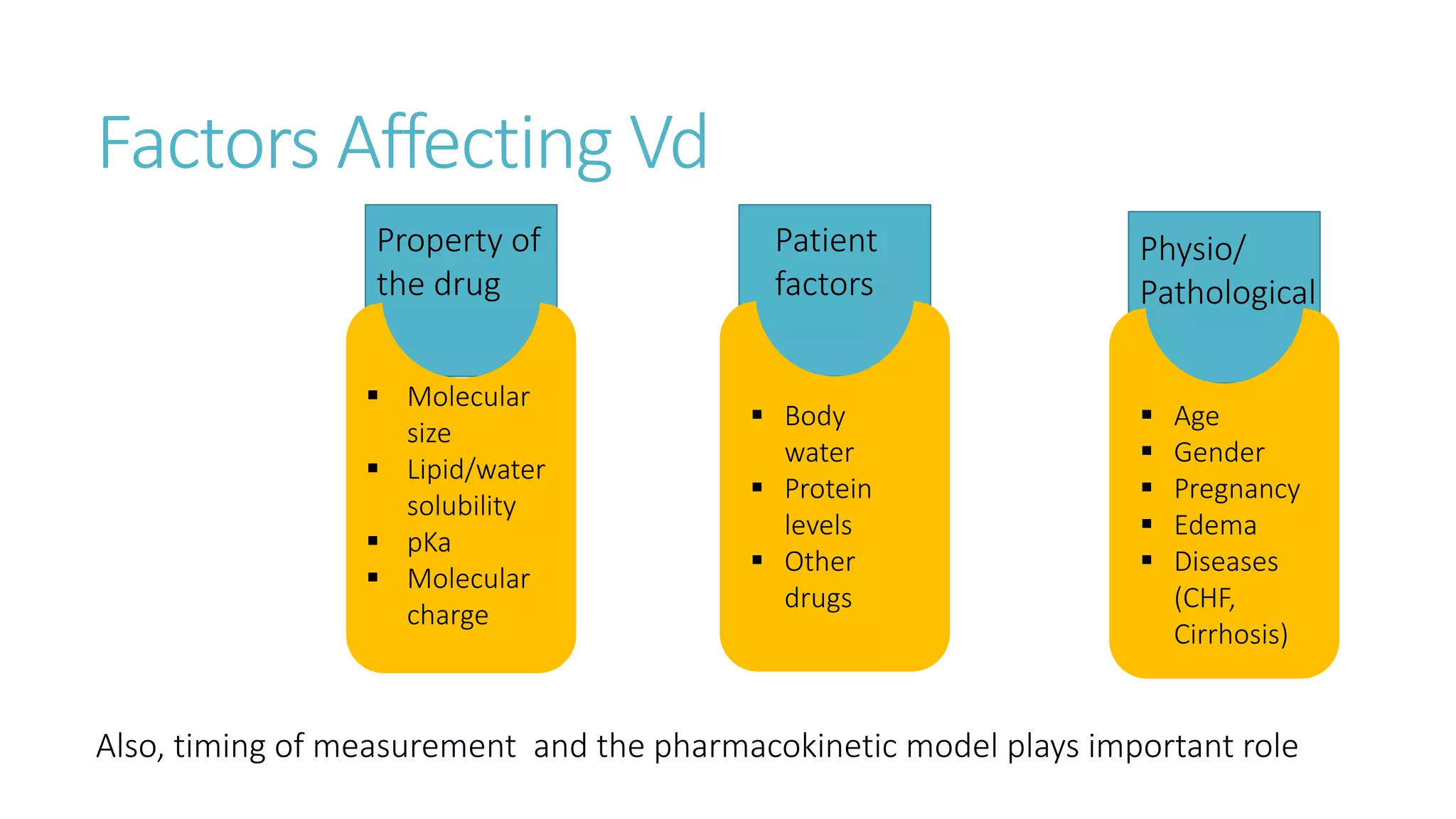
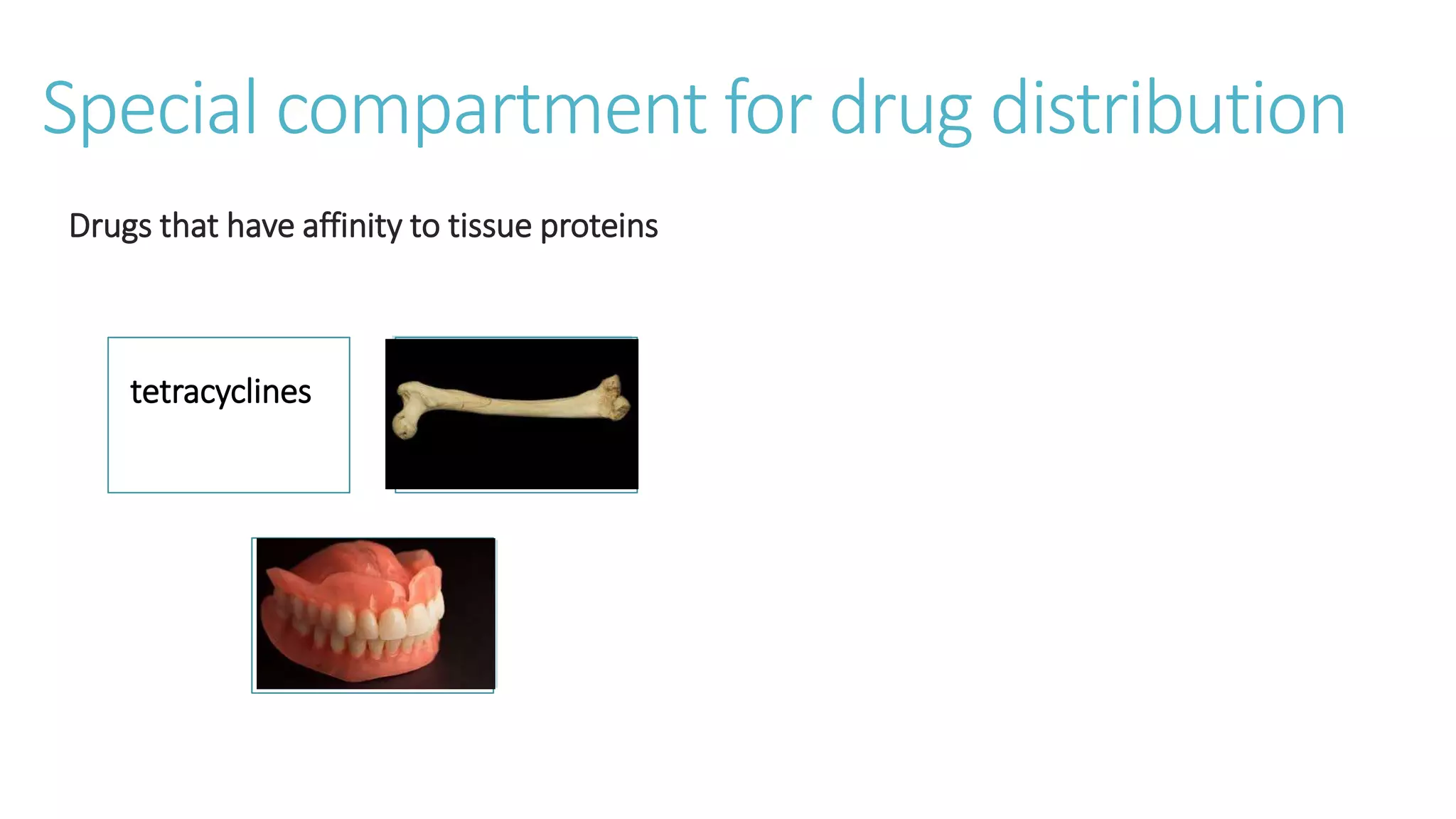
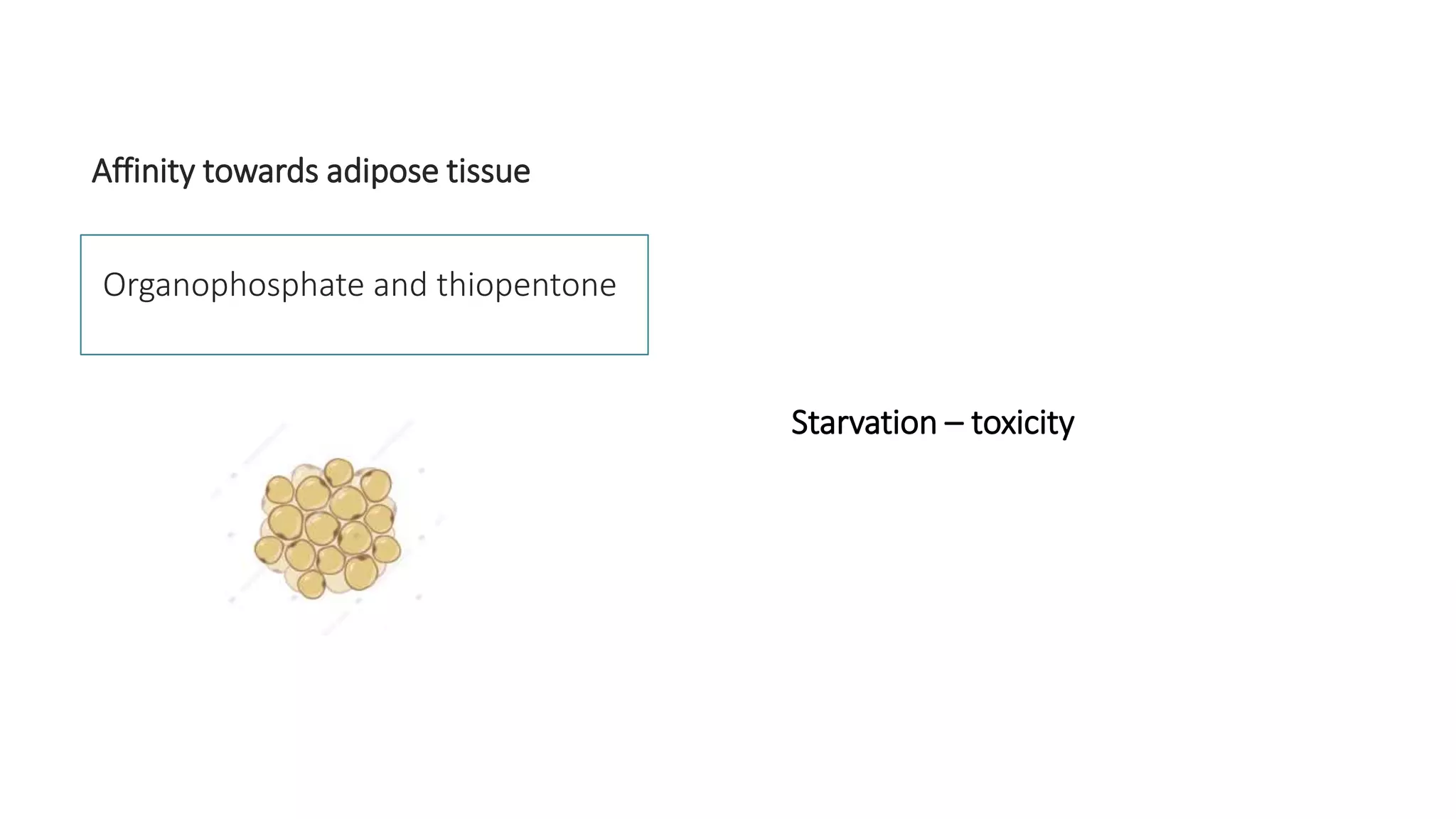
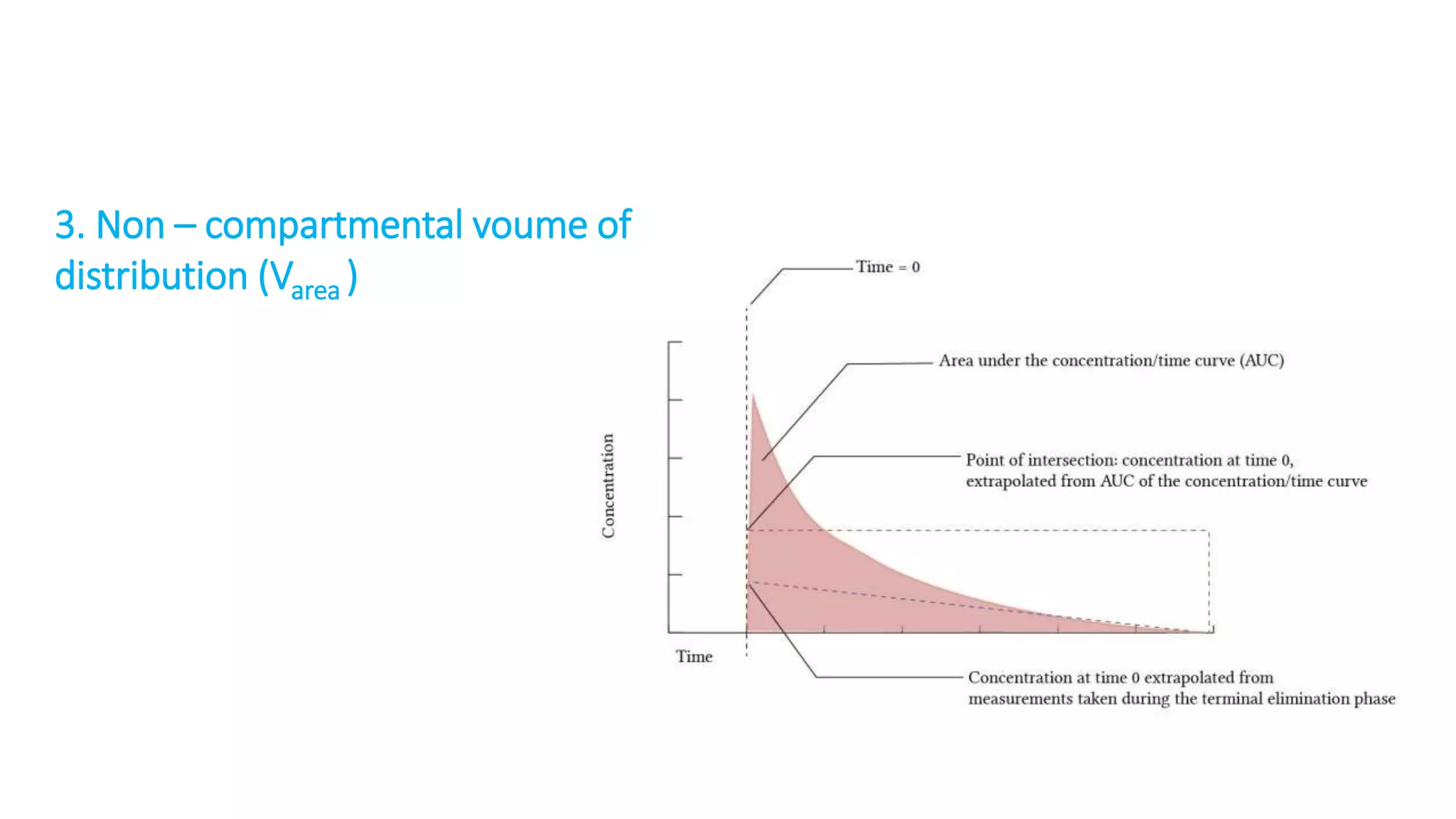
This document discusses volume of distribution (Vd), which is the apparent volume required to achieve a desired drug concentration in the body. It provides examples of calculating Vd based on drug dose and desired concentration in a fish tank or human patient. Factors that influence Vd include drug properties, patient characteristics, and physiological/pathological conditions. Vd can be single or multiple compartments, and its clinical significance includes determining loading doses and differences between pediatric/adult or obese/normal dosing. Special tissue compartments and redistribution are also discussed. In conclusion, understanding Vd aids in dosing drugs that require loading doses or are affected by protein binding and tissue storage.