Downloaded 2,113 times































































































The document discusses the preparation of compliant eCTD submissions, emphasizing the importance of content, process, standards, and technology for success. It outlines a case study involving a small biotech company that faced challenges during its NDA submission to the FDA, including a 'refuse-to-file' designation due to compliance issues. Key factors are highlighted, such as the significance of accurate file organization and the understanding of regulatory requirements for eCTD submissions.
Introduction to compliant eCTD submissions and agenda overview.
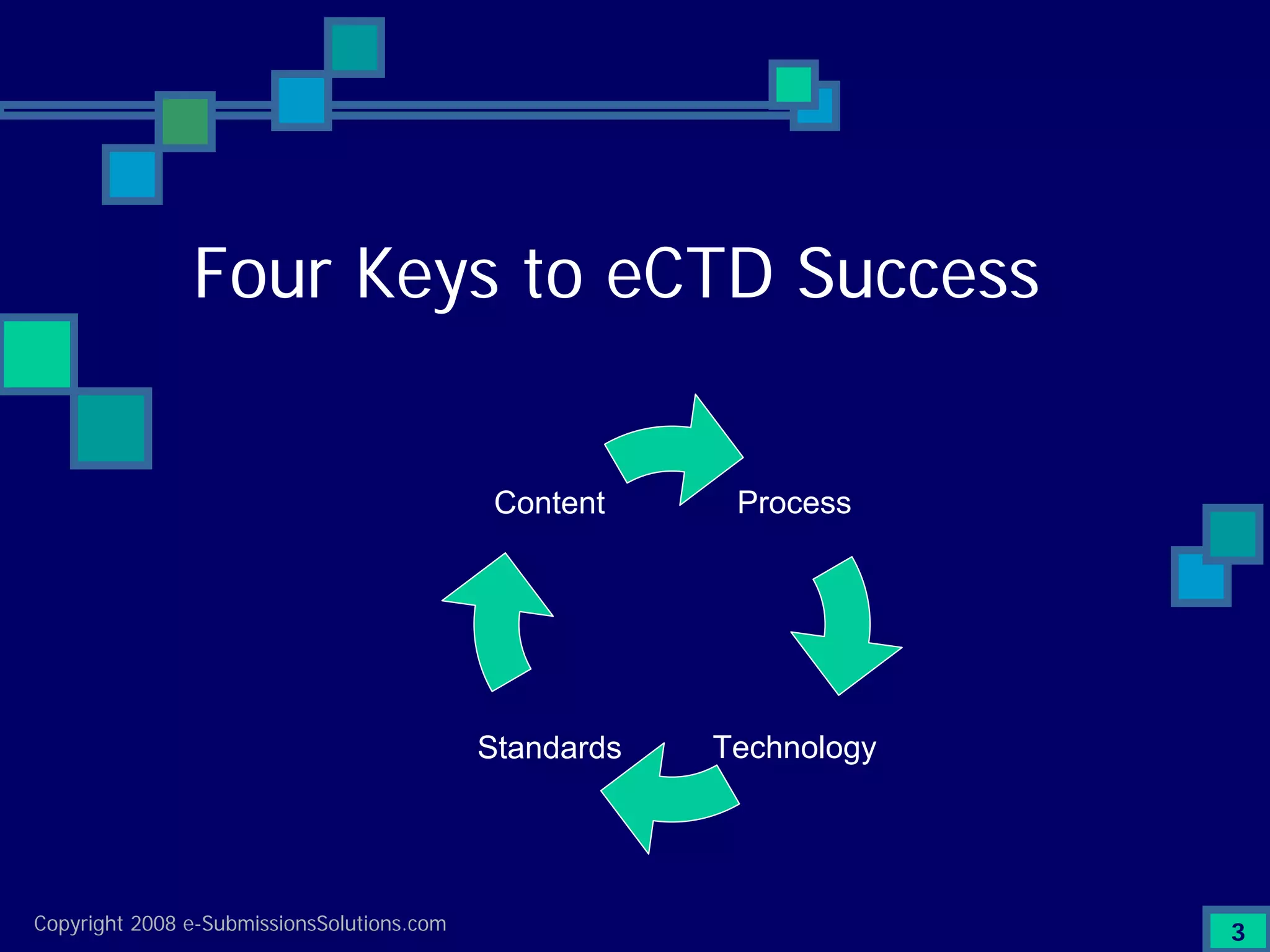
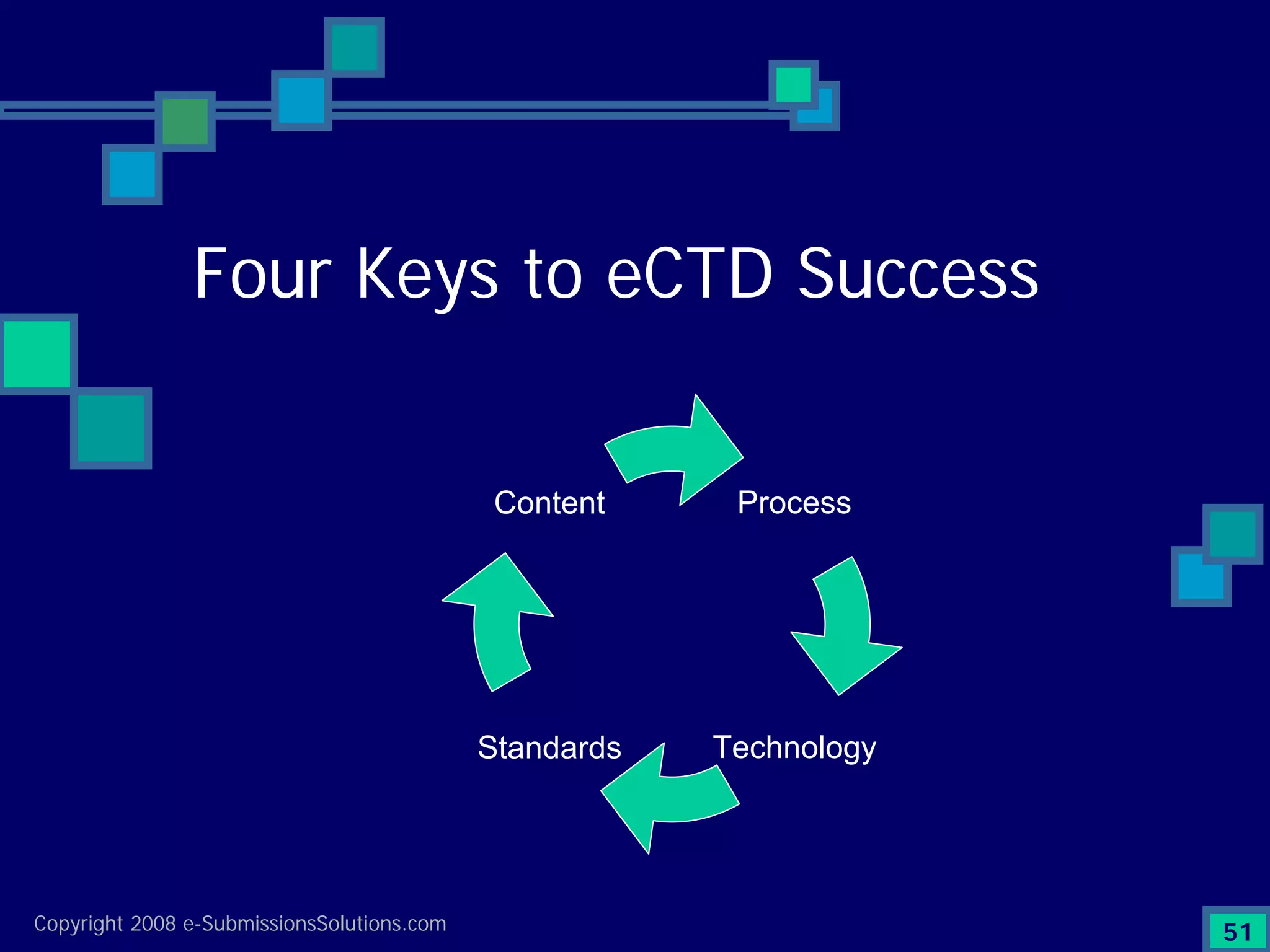
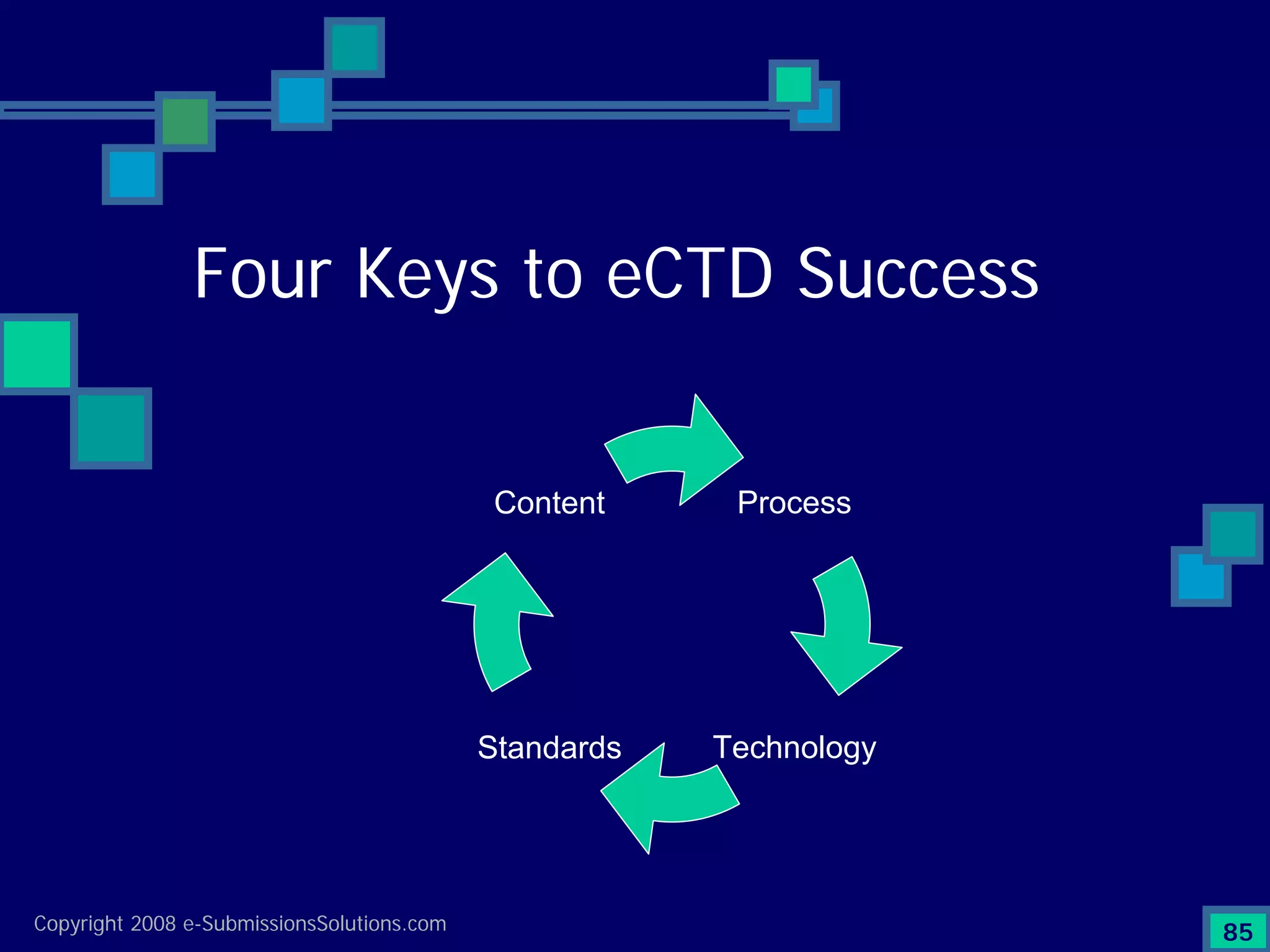
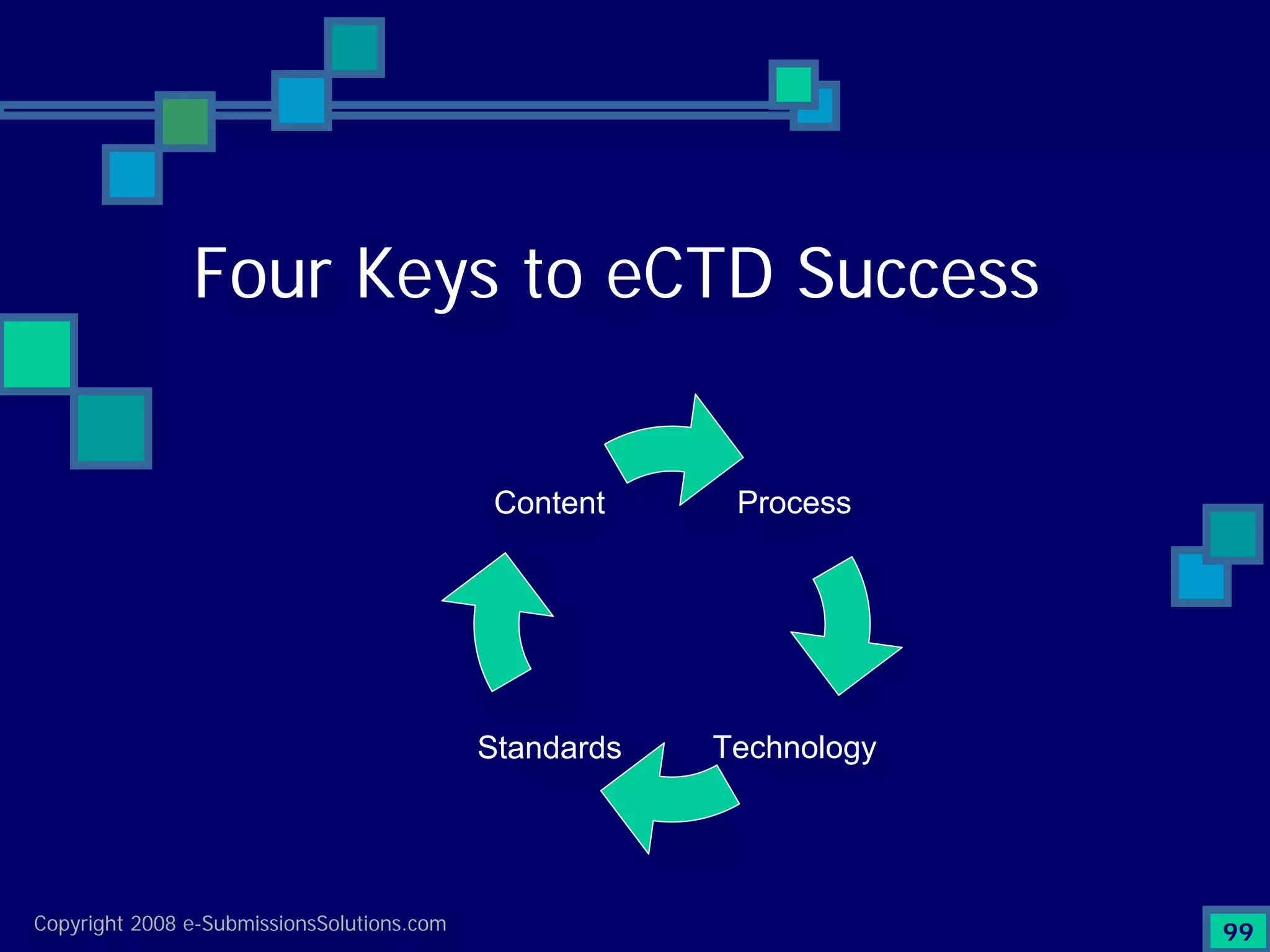
Discusses Content, Process, Standards, and Technology as crucial factors for successful eCTD.
Details a case study facing a biotech's NDA submission issues with RTF by FDA, impacting partnerships & workforce.
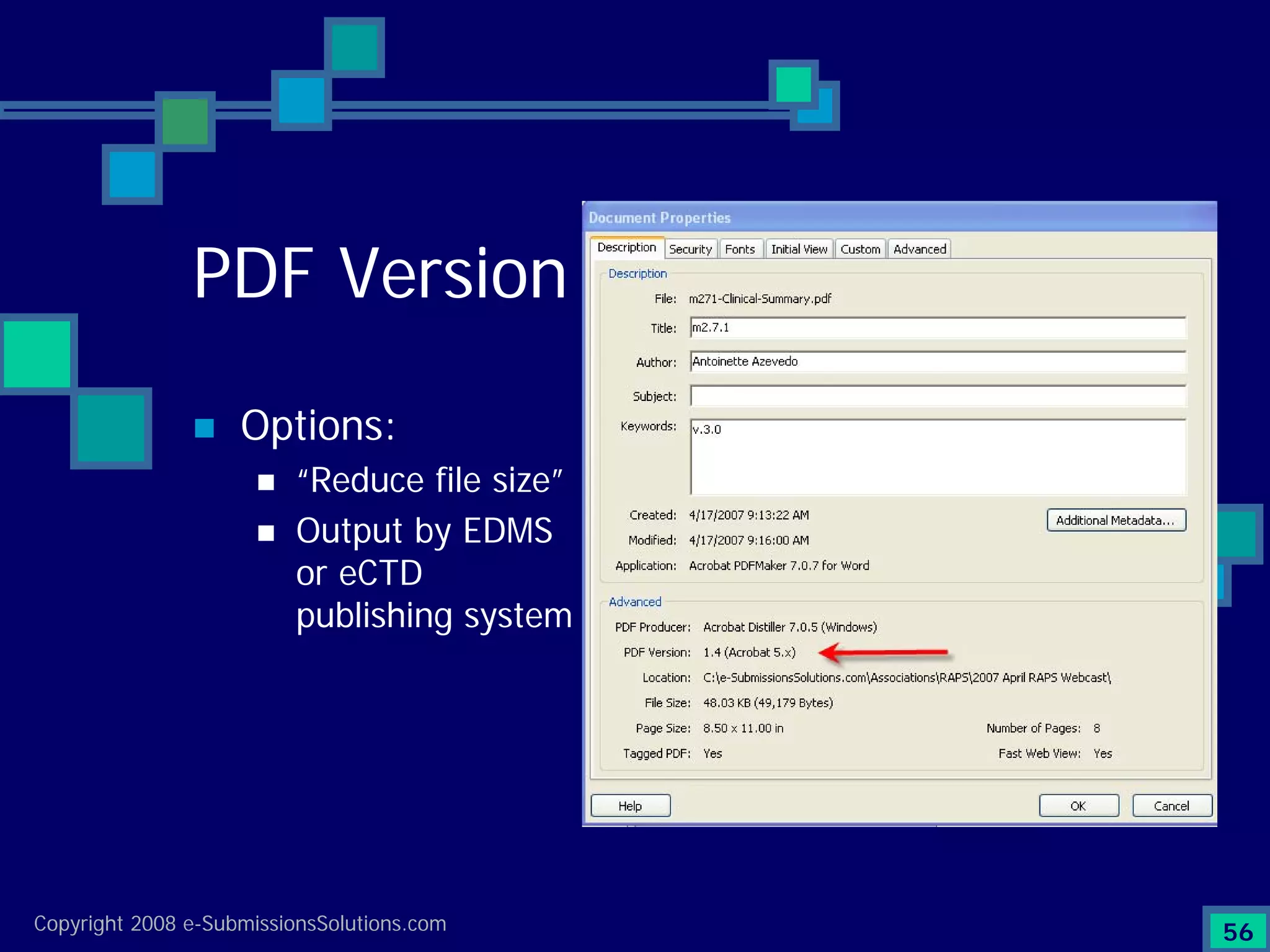
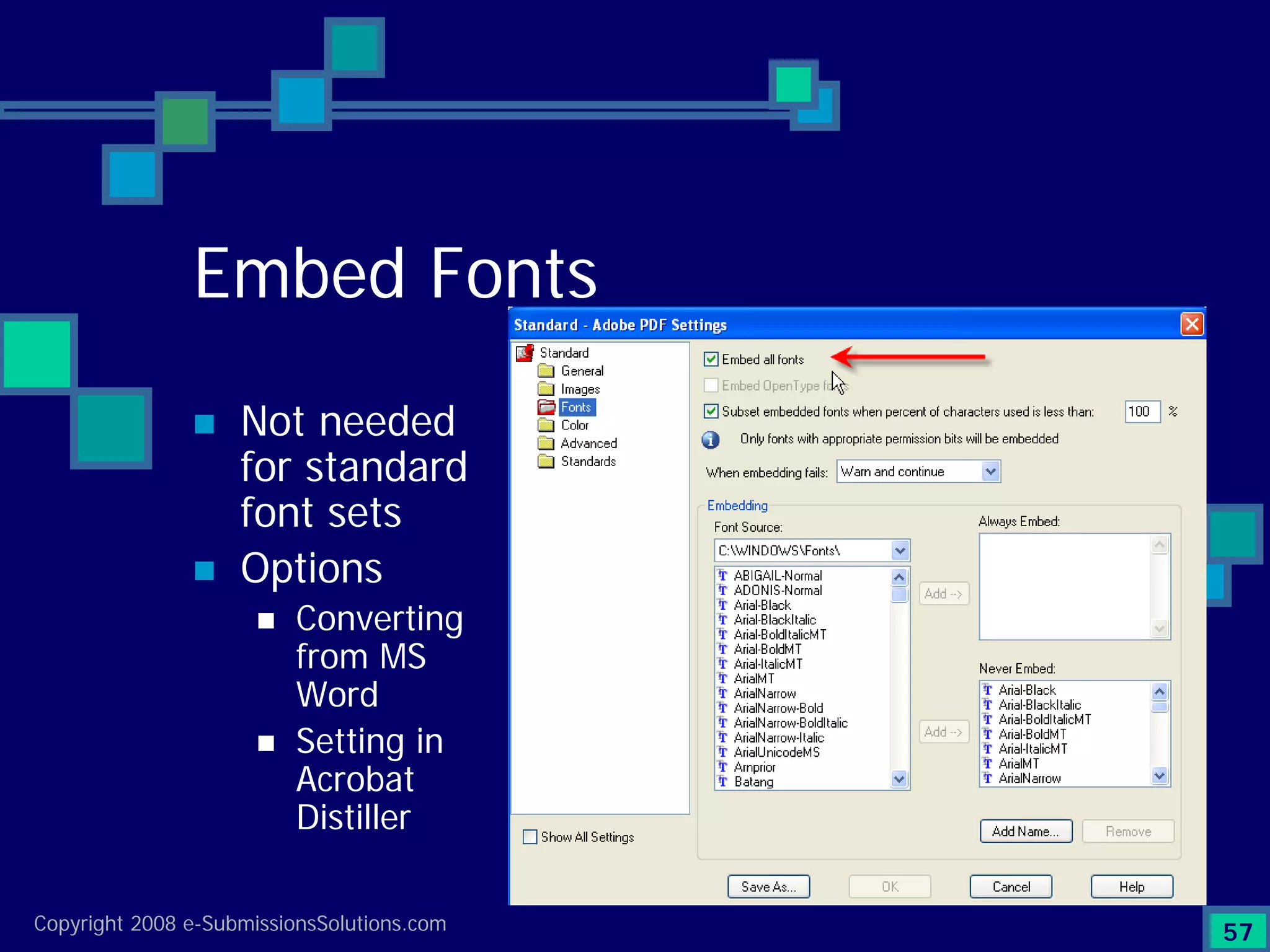
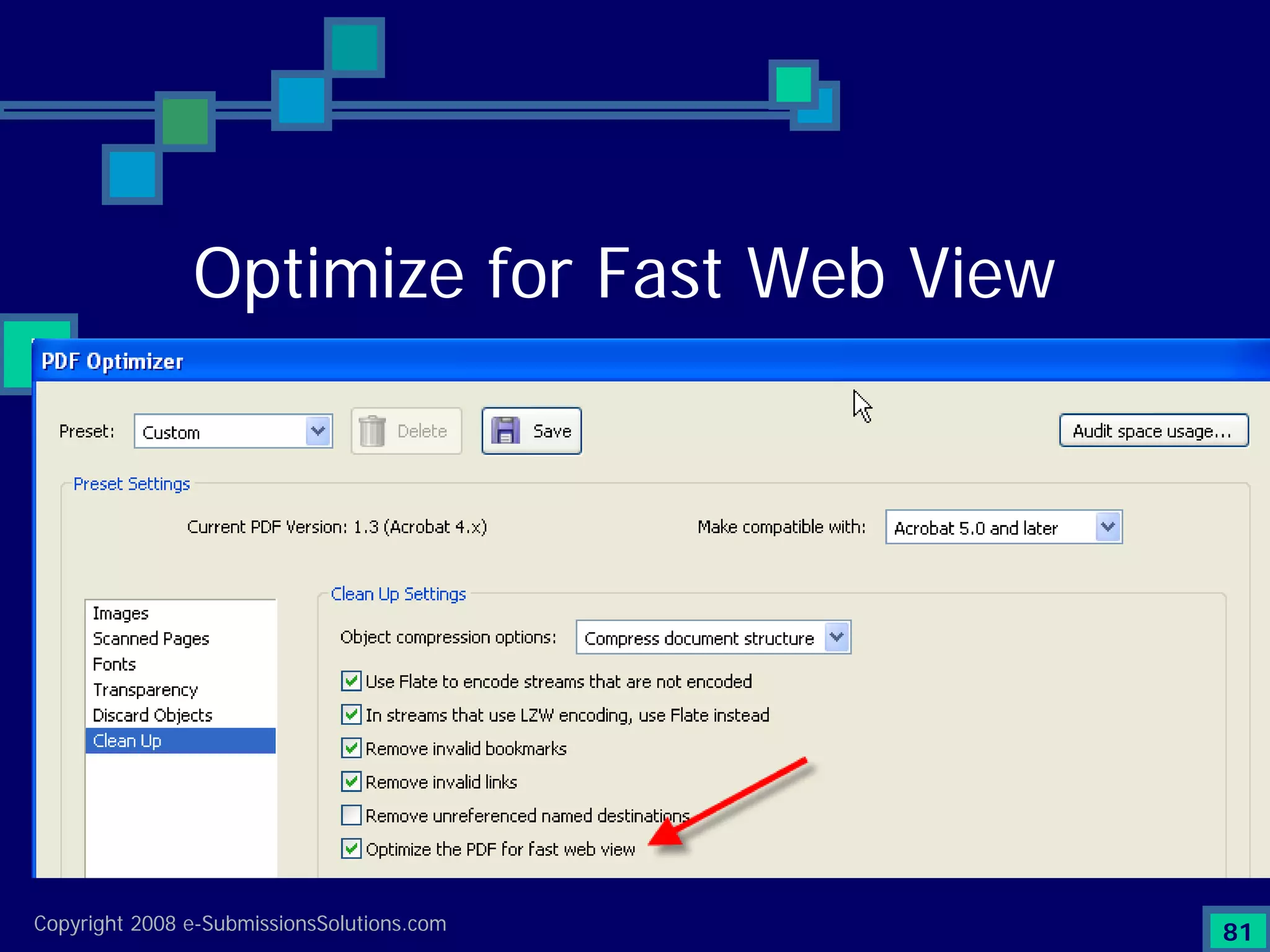
Identifies top technical issues and compliance challenges in eCTD submissions as per FDA.
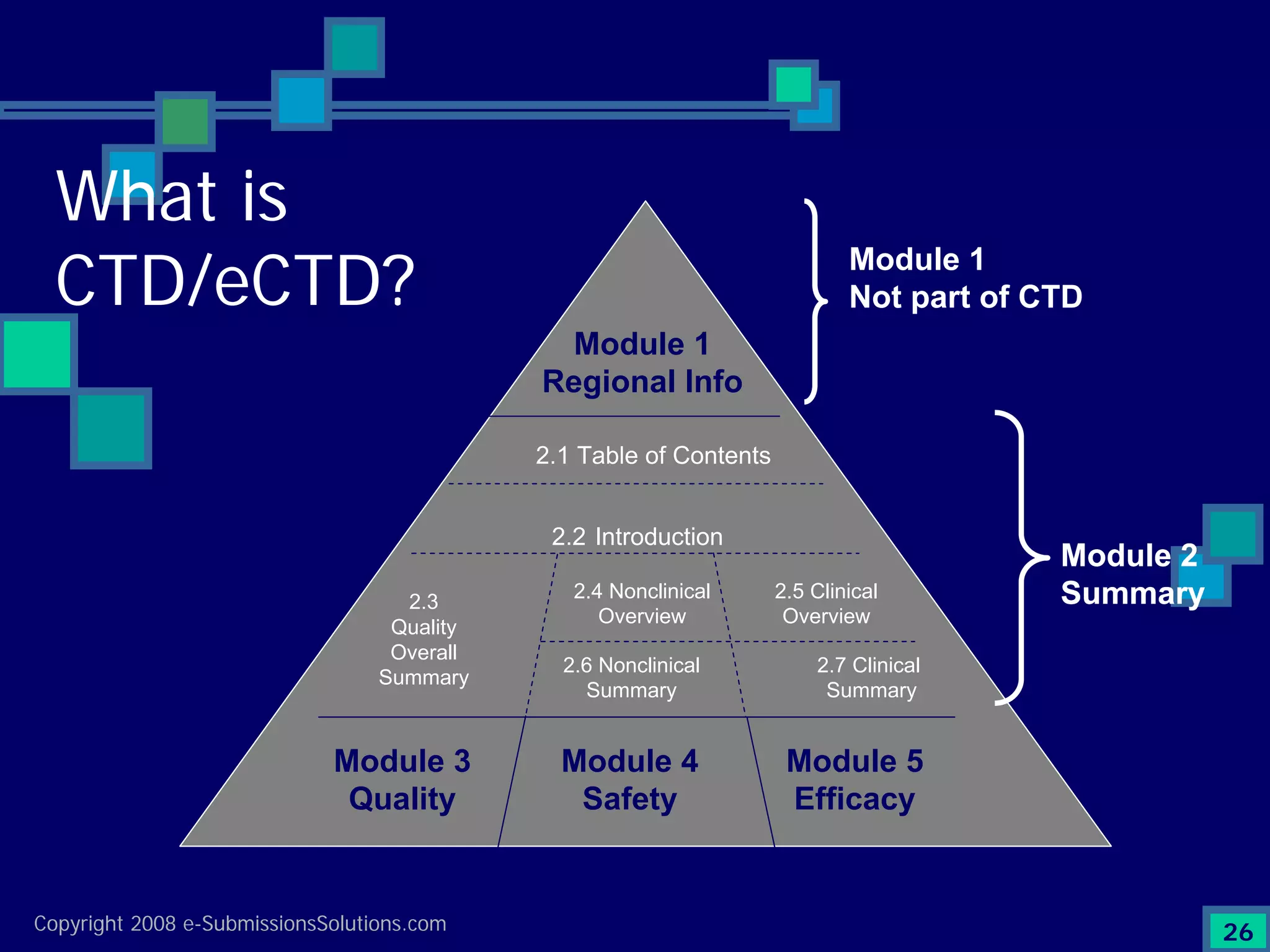
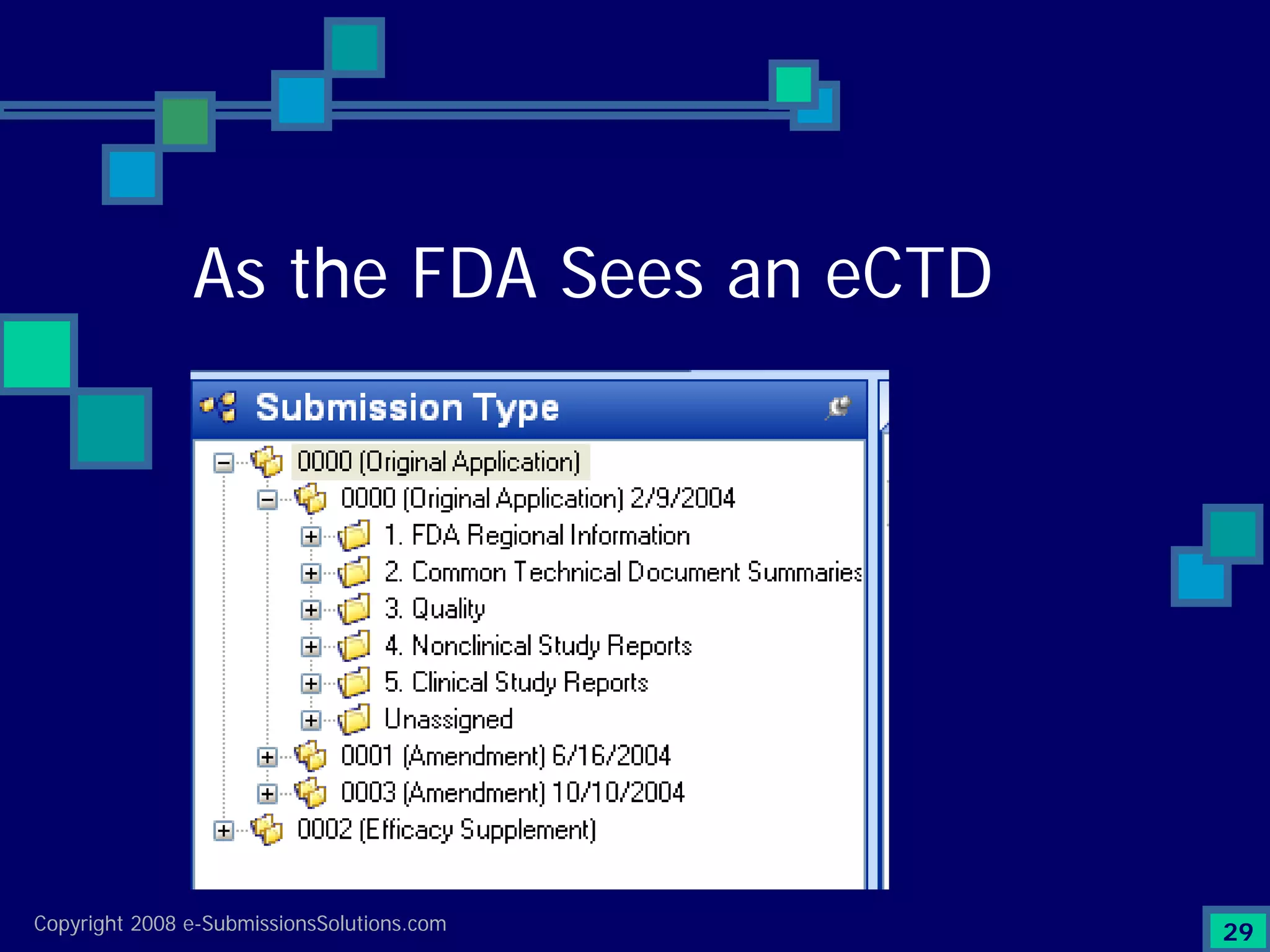
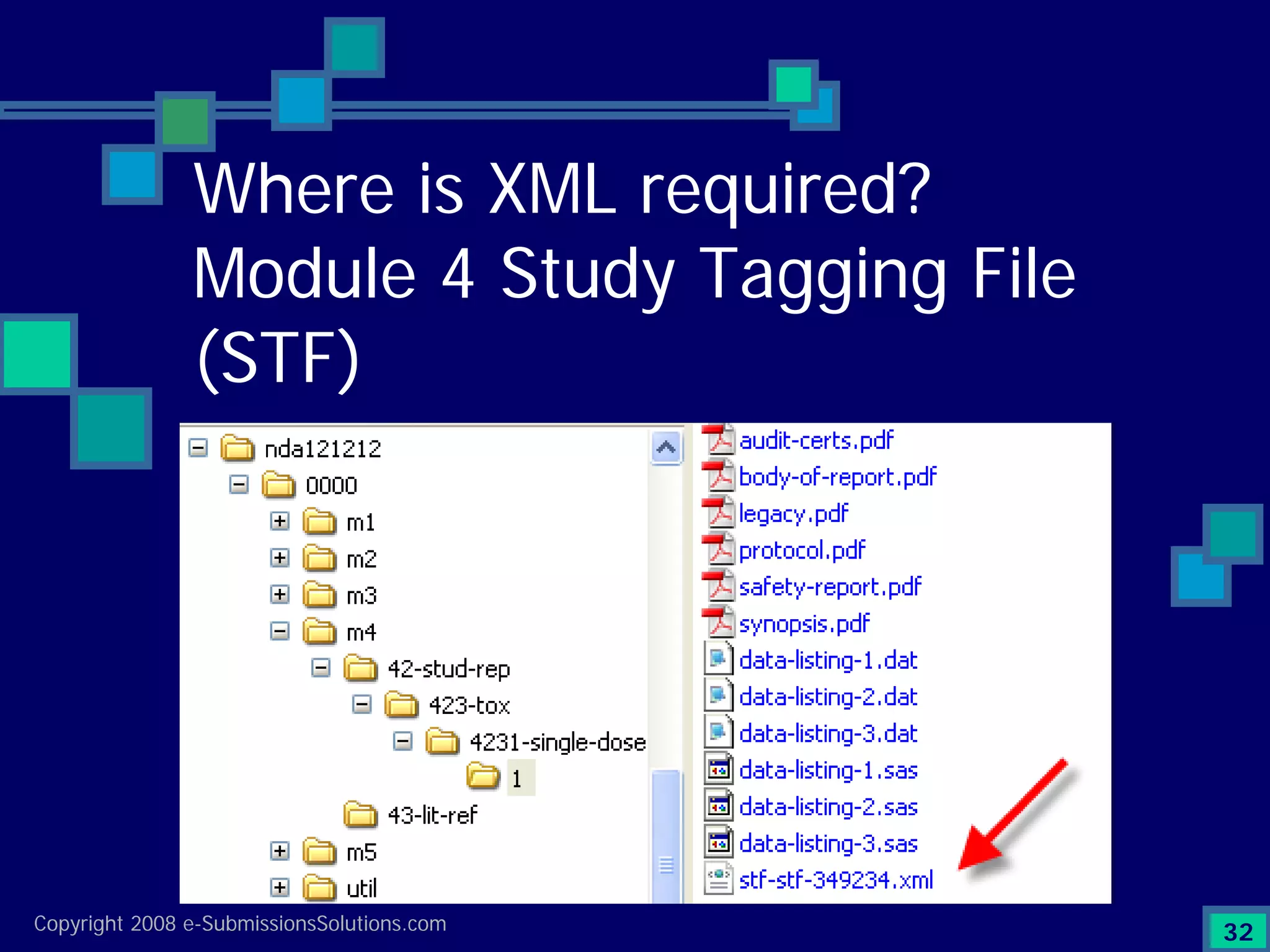
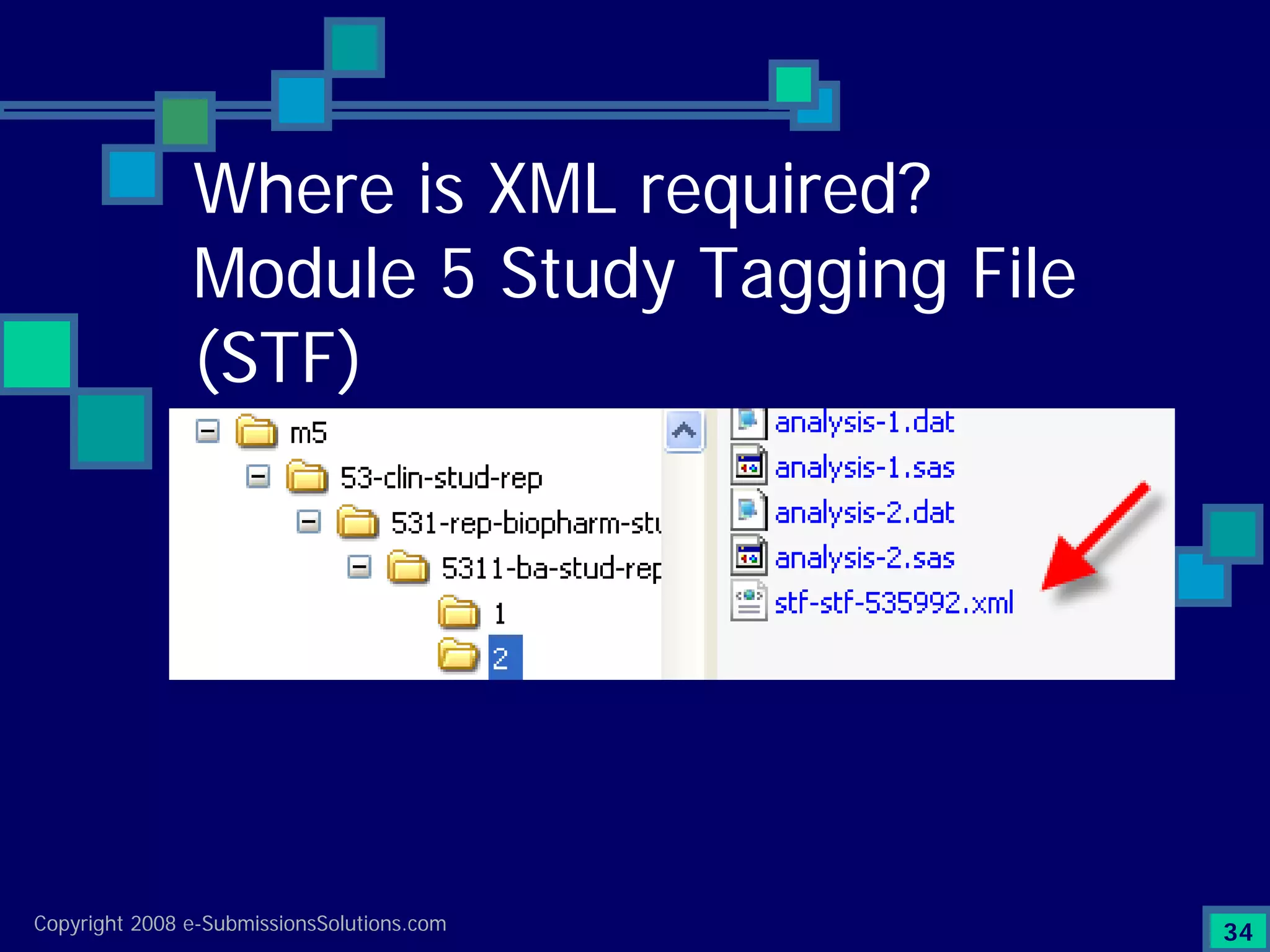
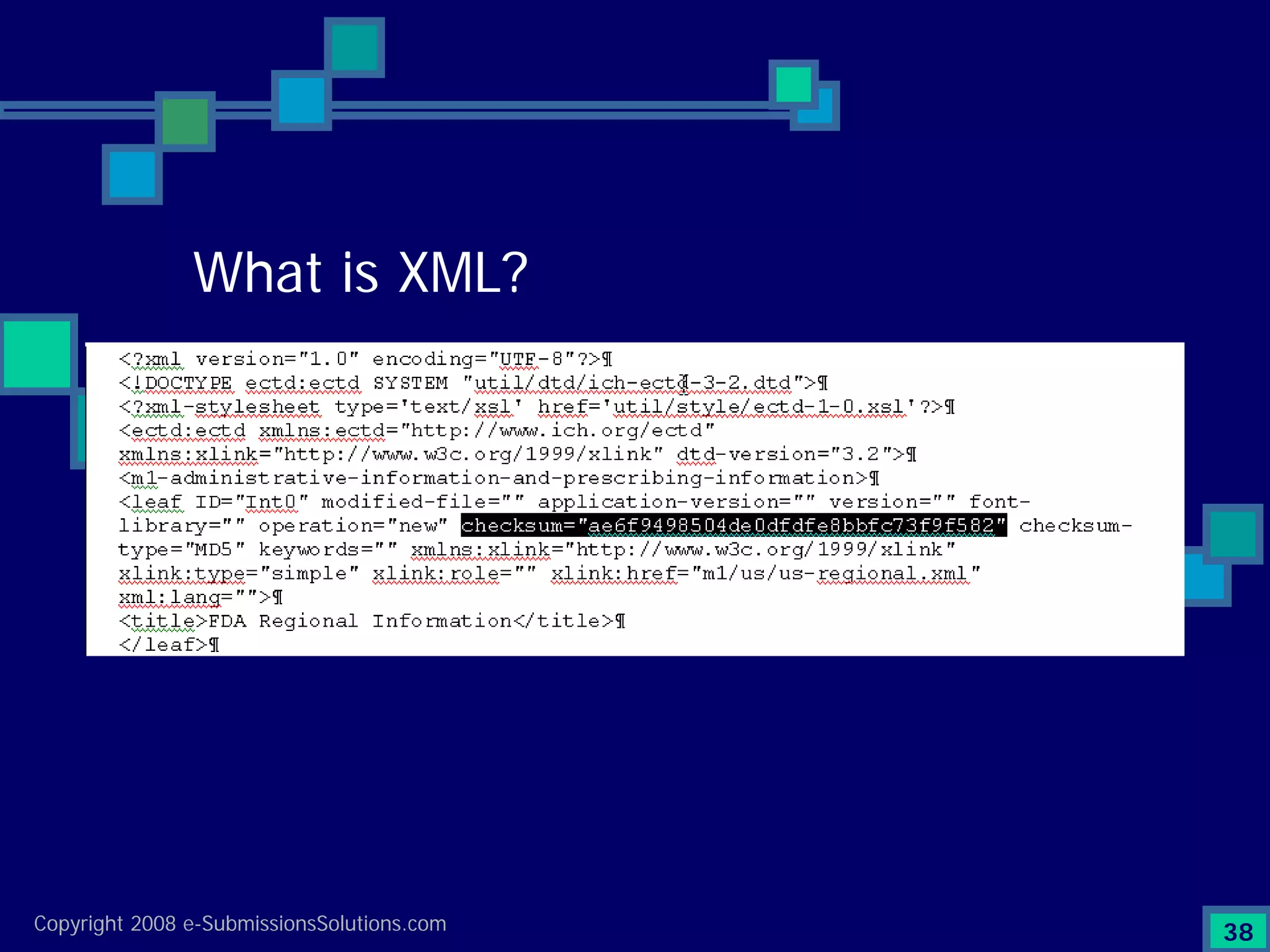
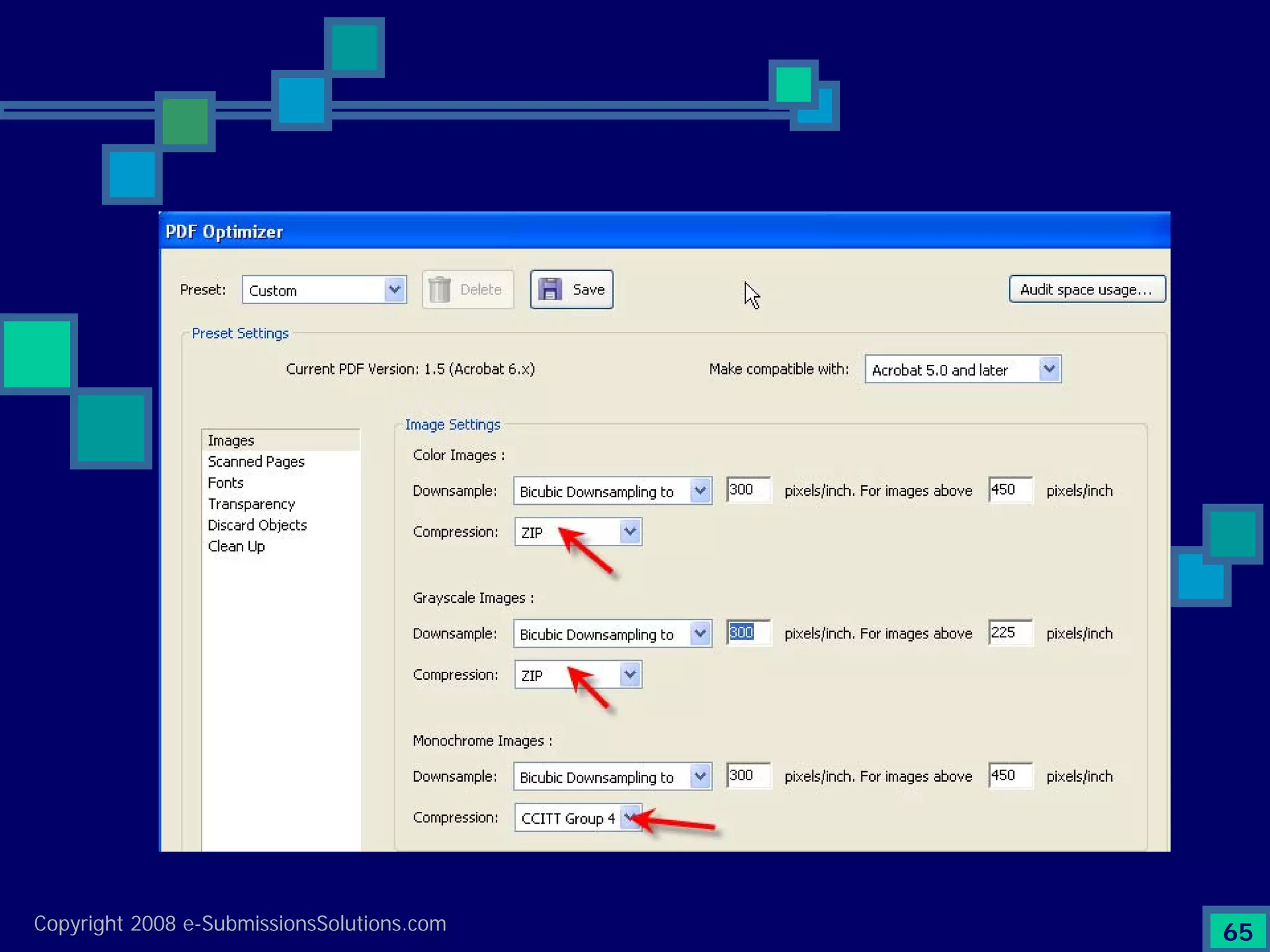
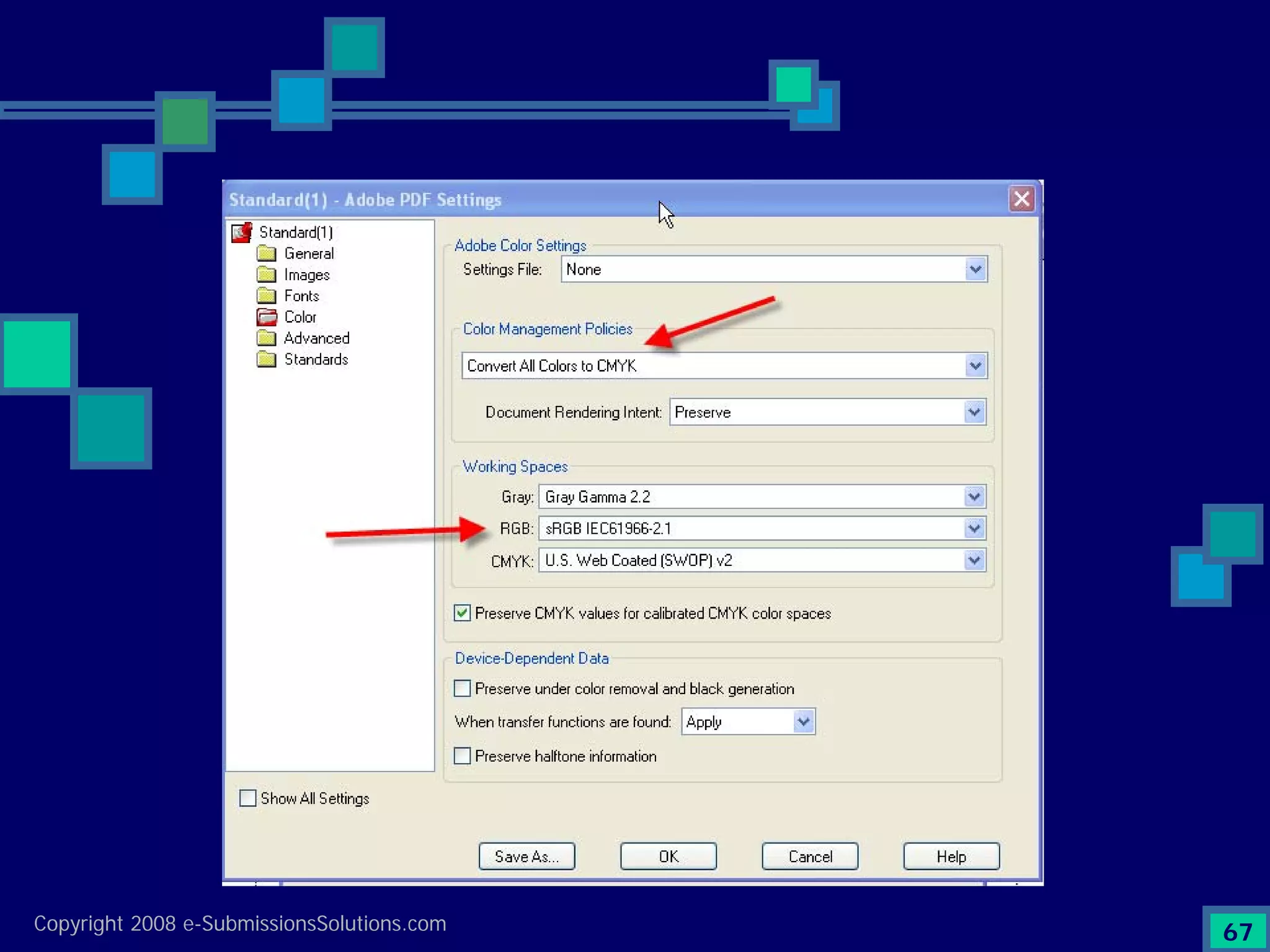
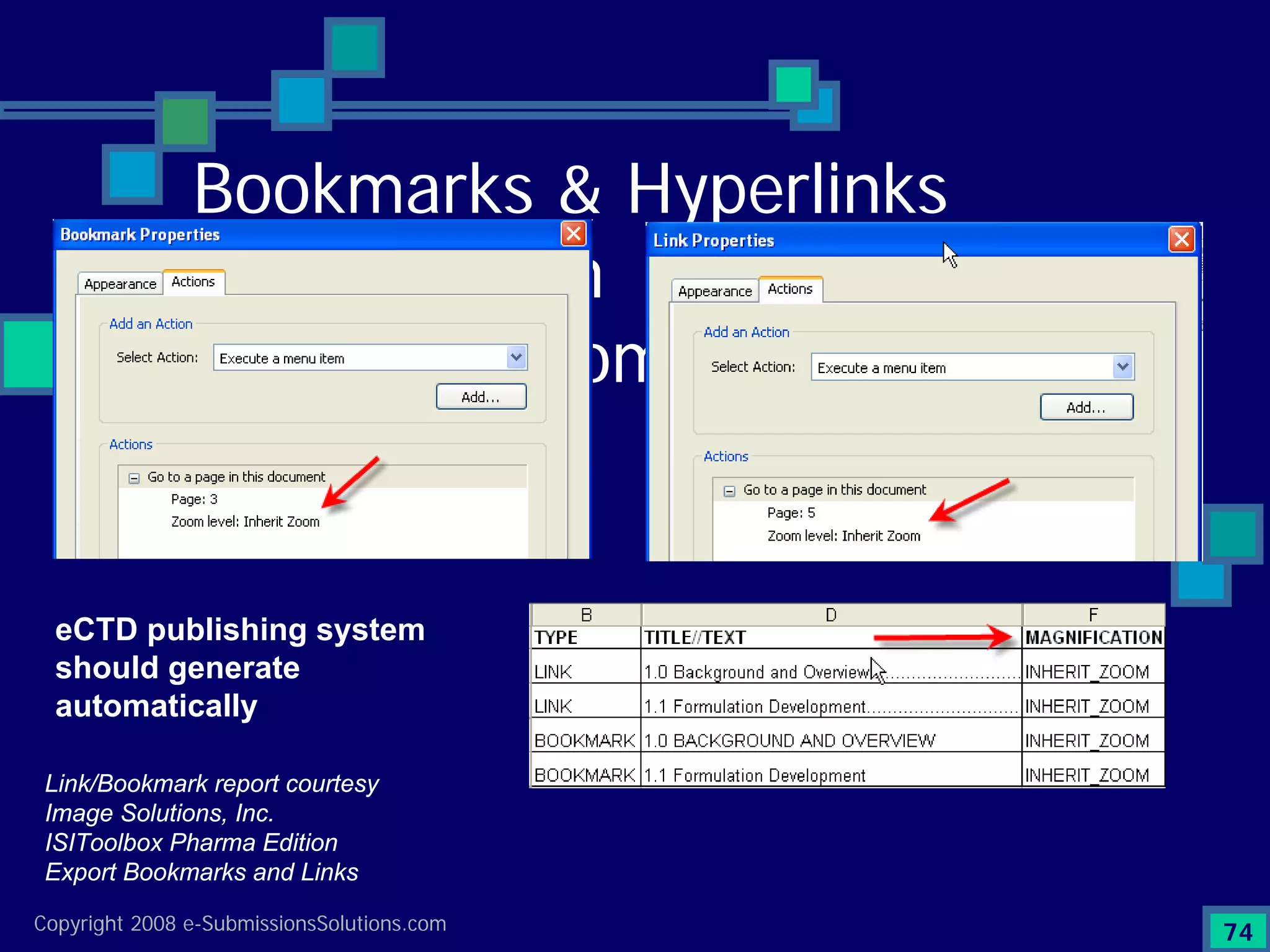
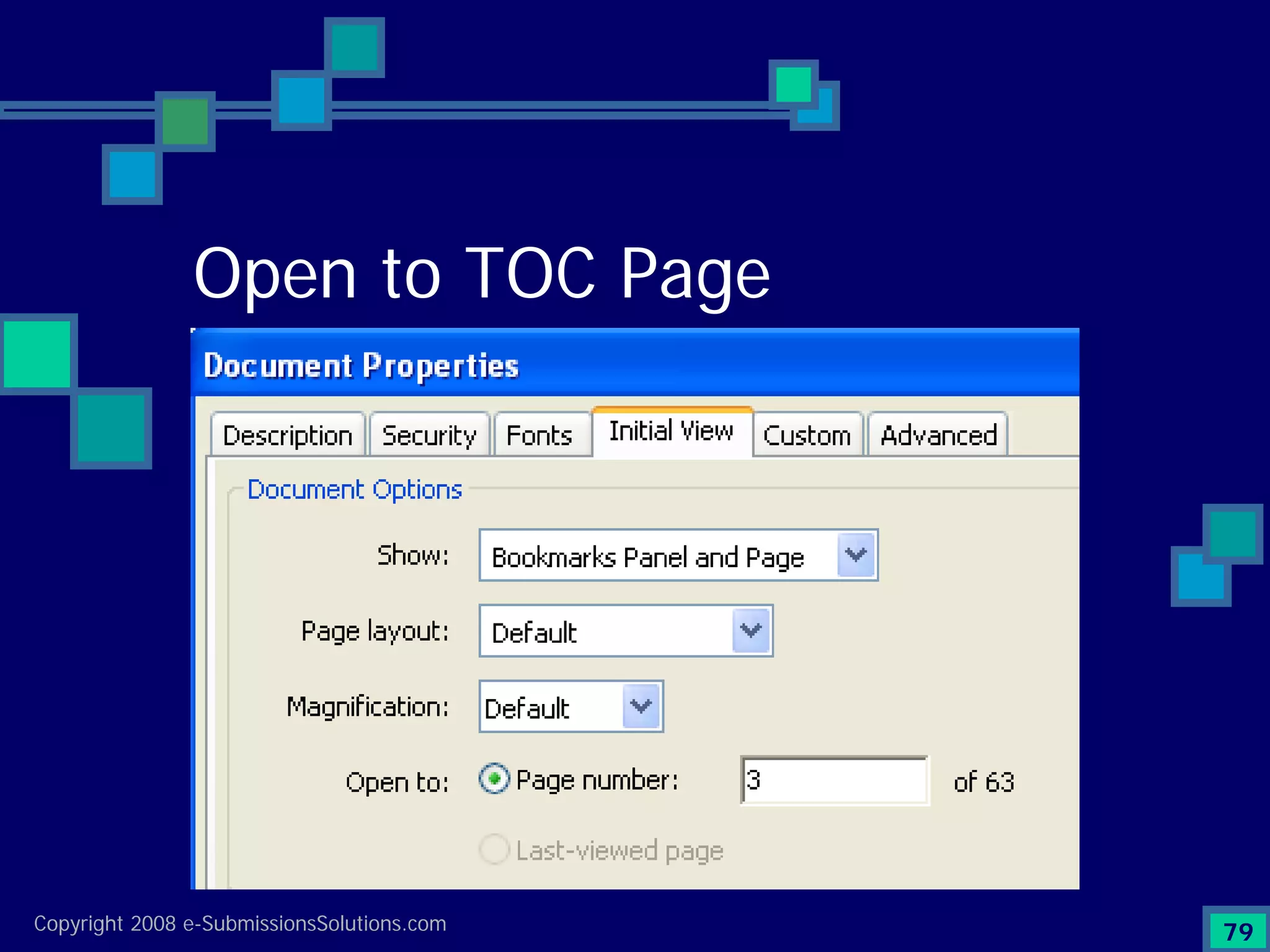
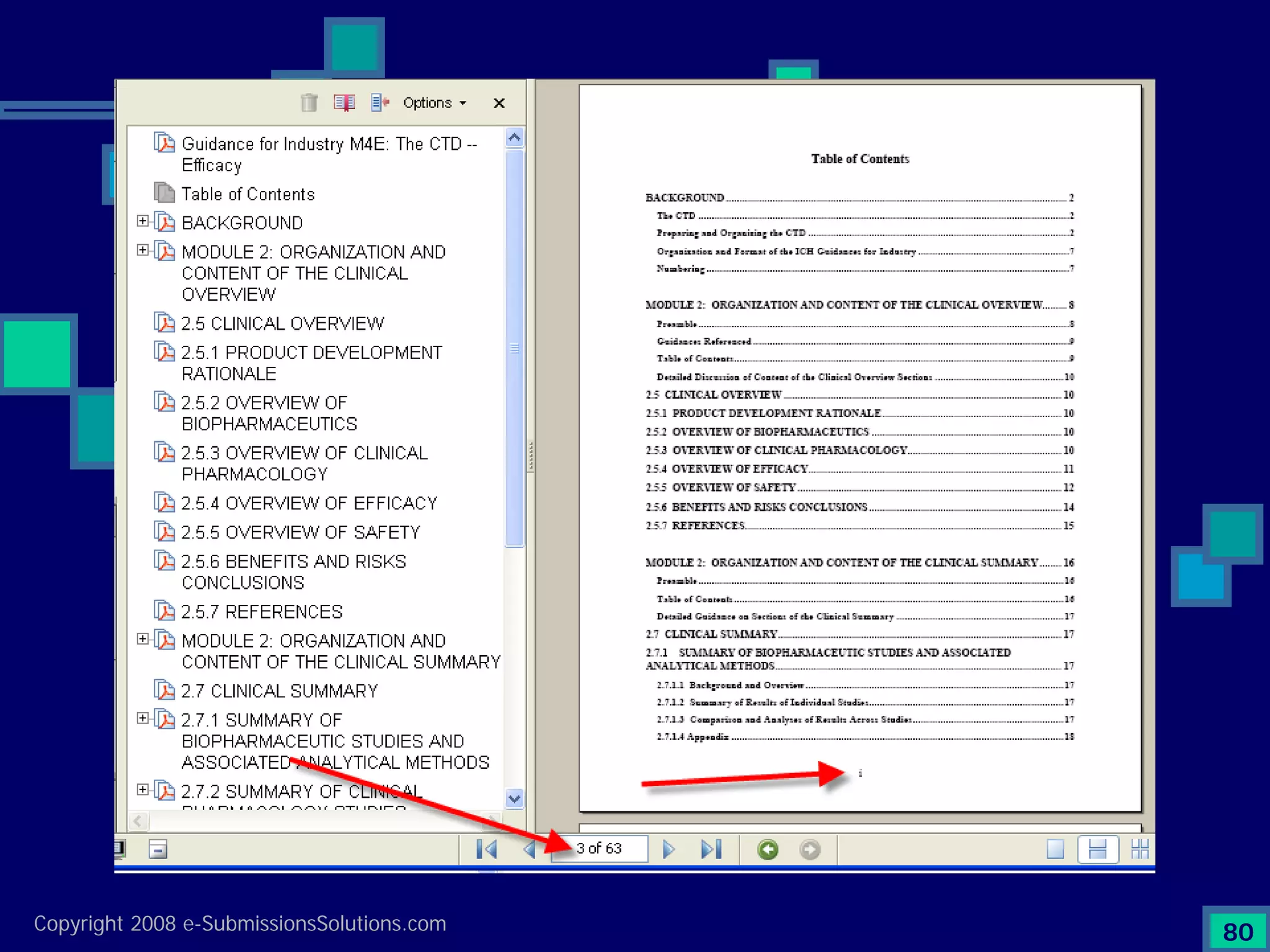
Explains CTD/eCTD modules including Module 1 for regional info and others for quality, safety, and efficacy. Discusses the use of XML structure for various submission modules and potential XML-related issues.
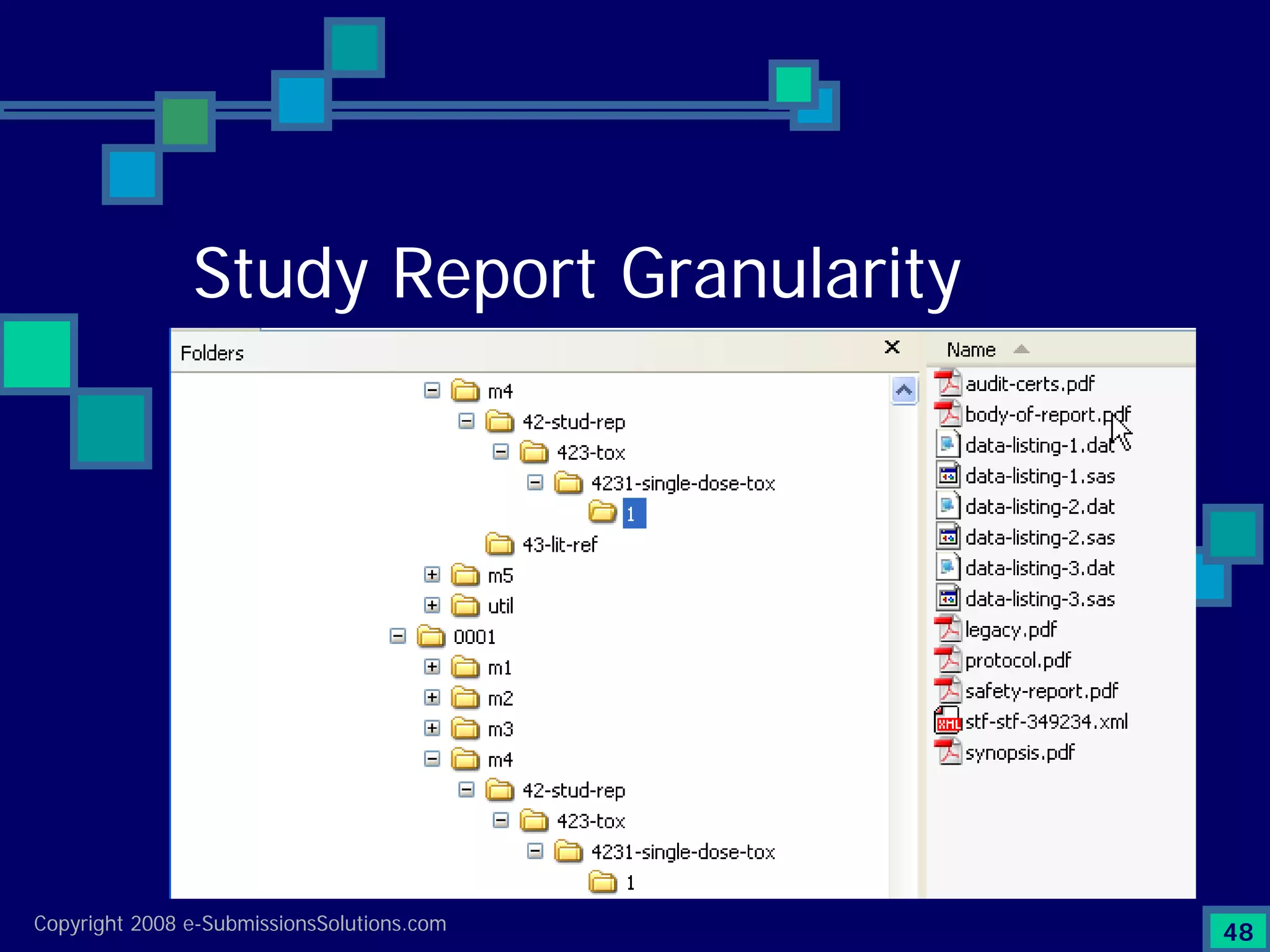
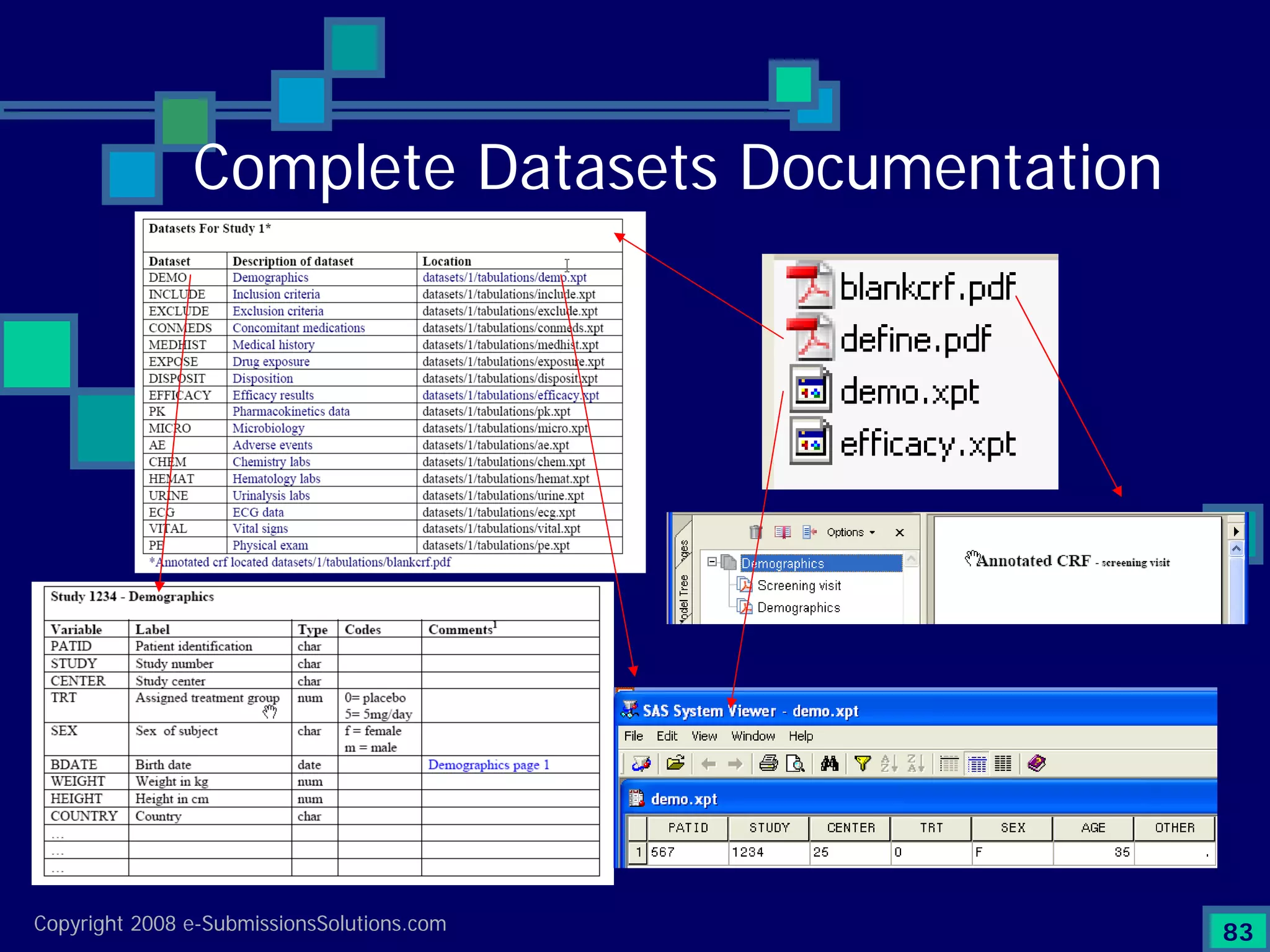
Documents challenges related to authoring, data granularity, collection and consistency from study sites.
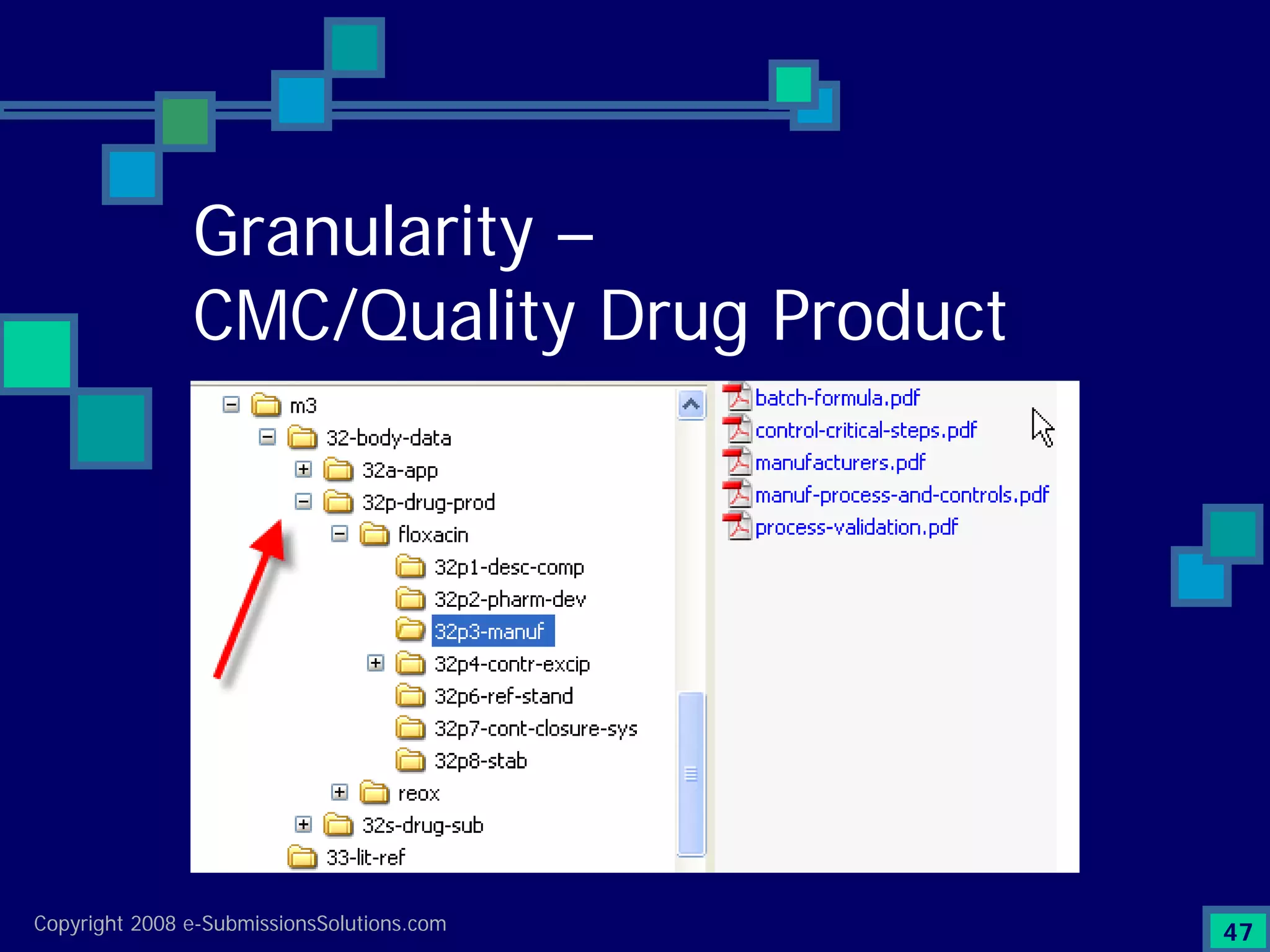
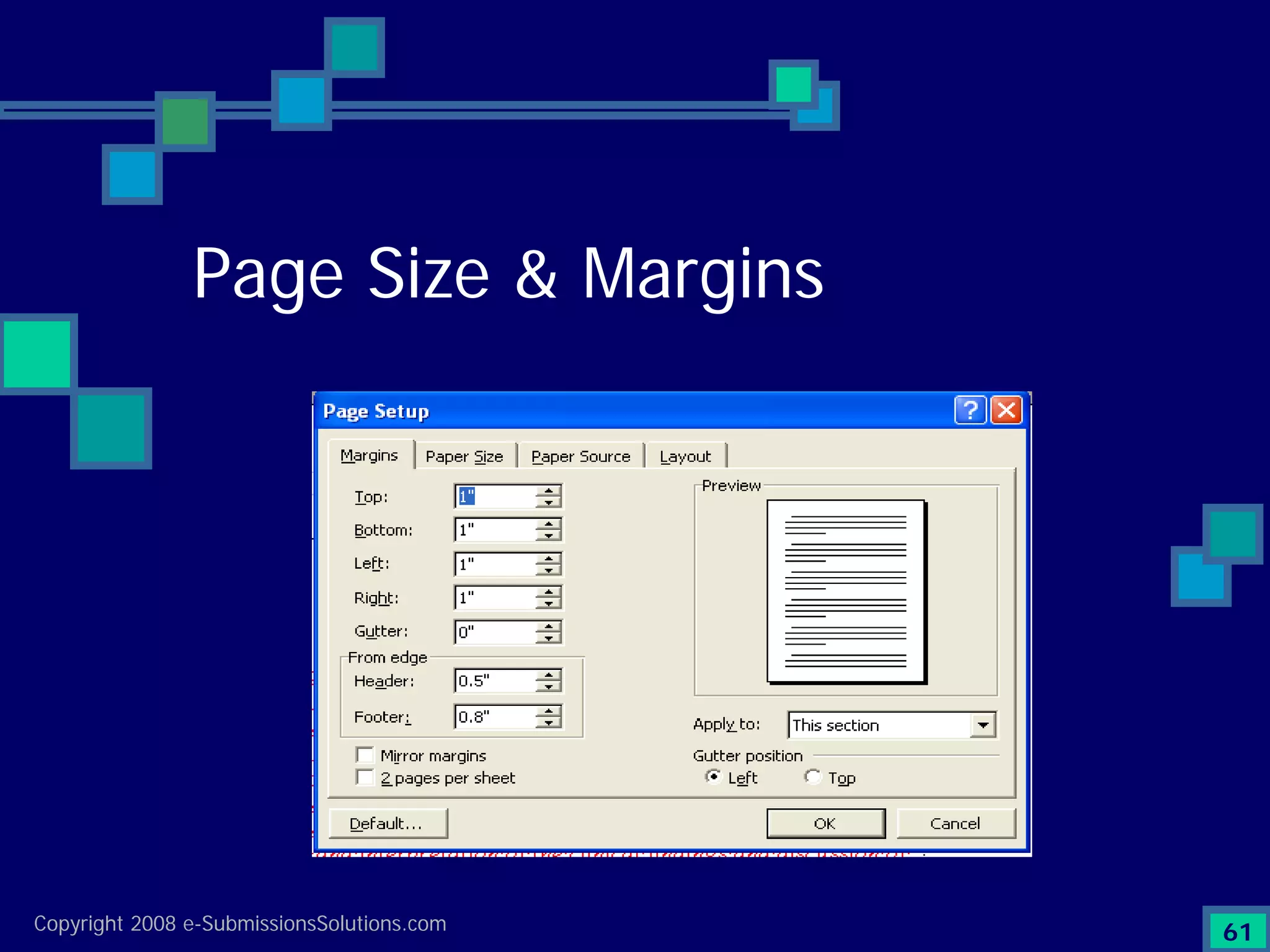
Emphasizes the granularity required in document organization and highlights the necessity for compliant submissions.
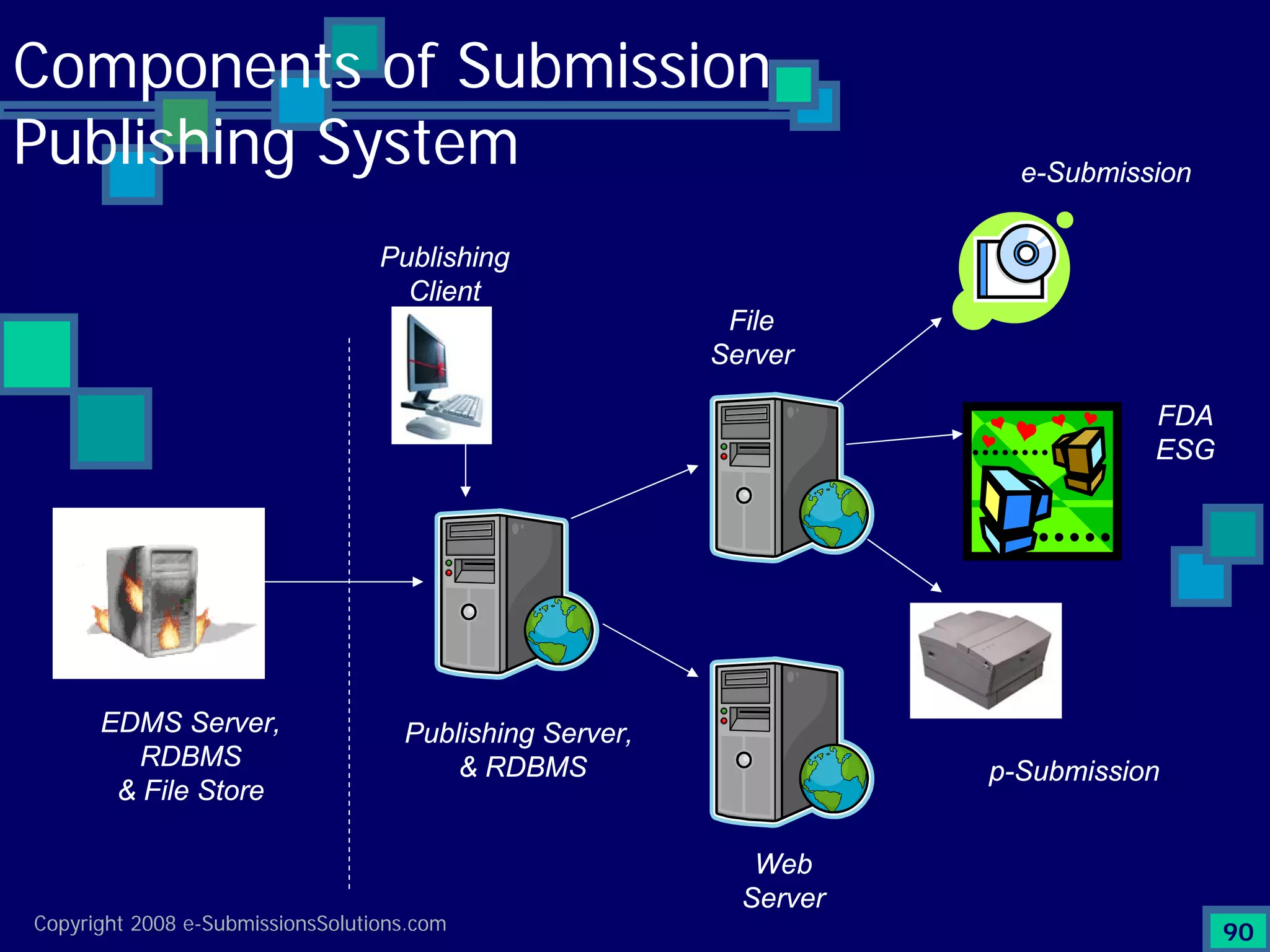
Details workflow from document collection and verification to the preparation of submissions and compliance.
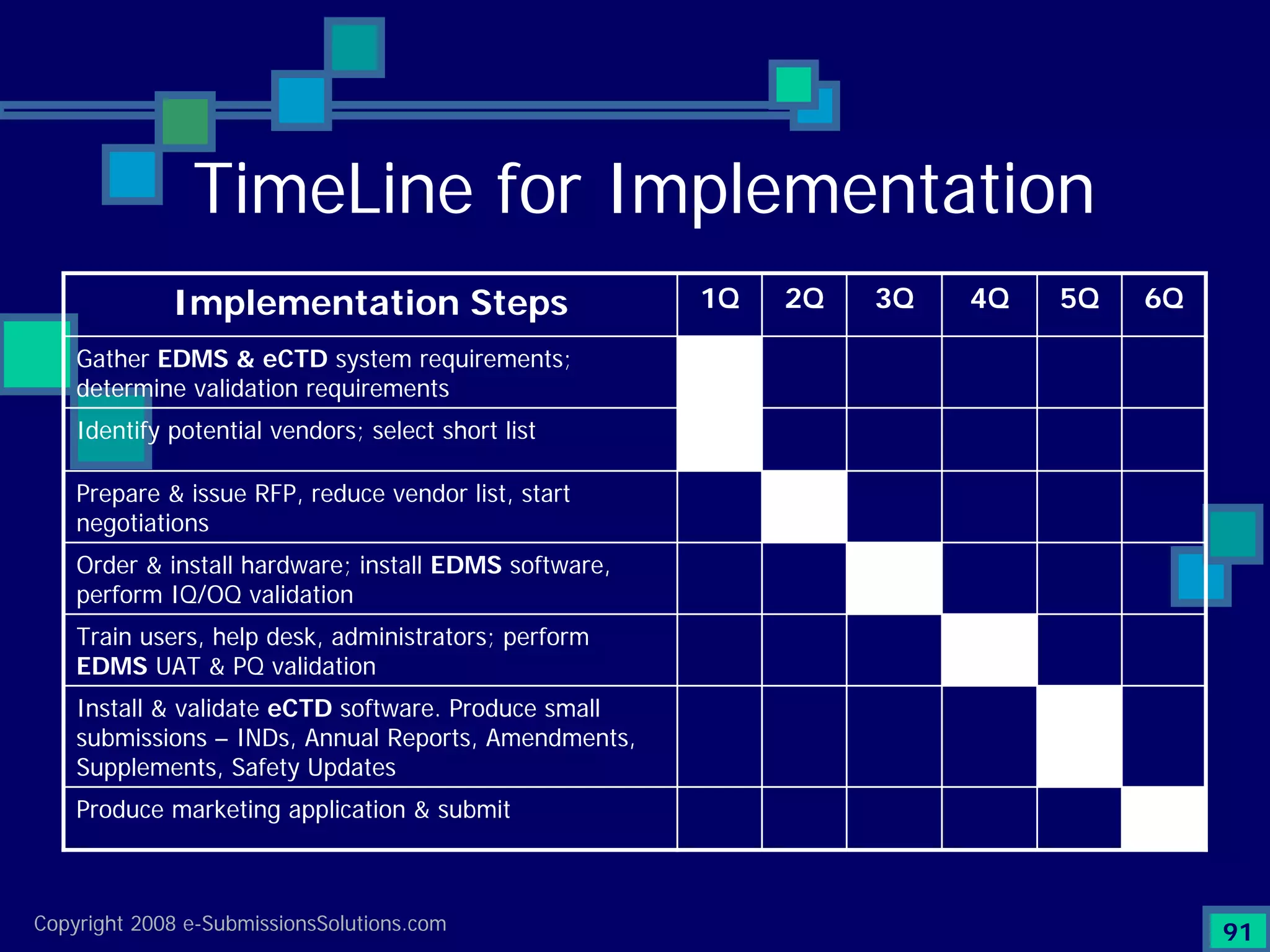
Analyzes both ROI for implementing eCTD systems and the associated costs benefits compared to traditional methods. Outlines current eCTD mandates and transitioning approaches in FDA, EU, Canada, Japan and summarizes progress.Explains FDA's strategy for implementing electronic submissions and the evolving regulatory landscape.
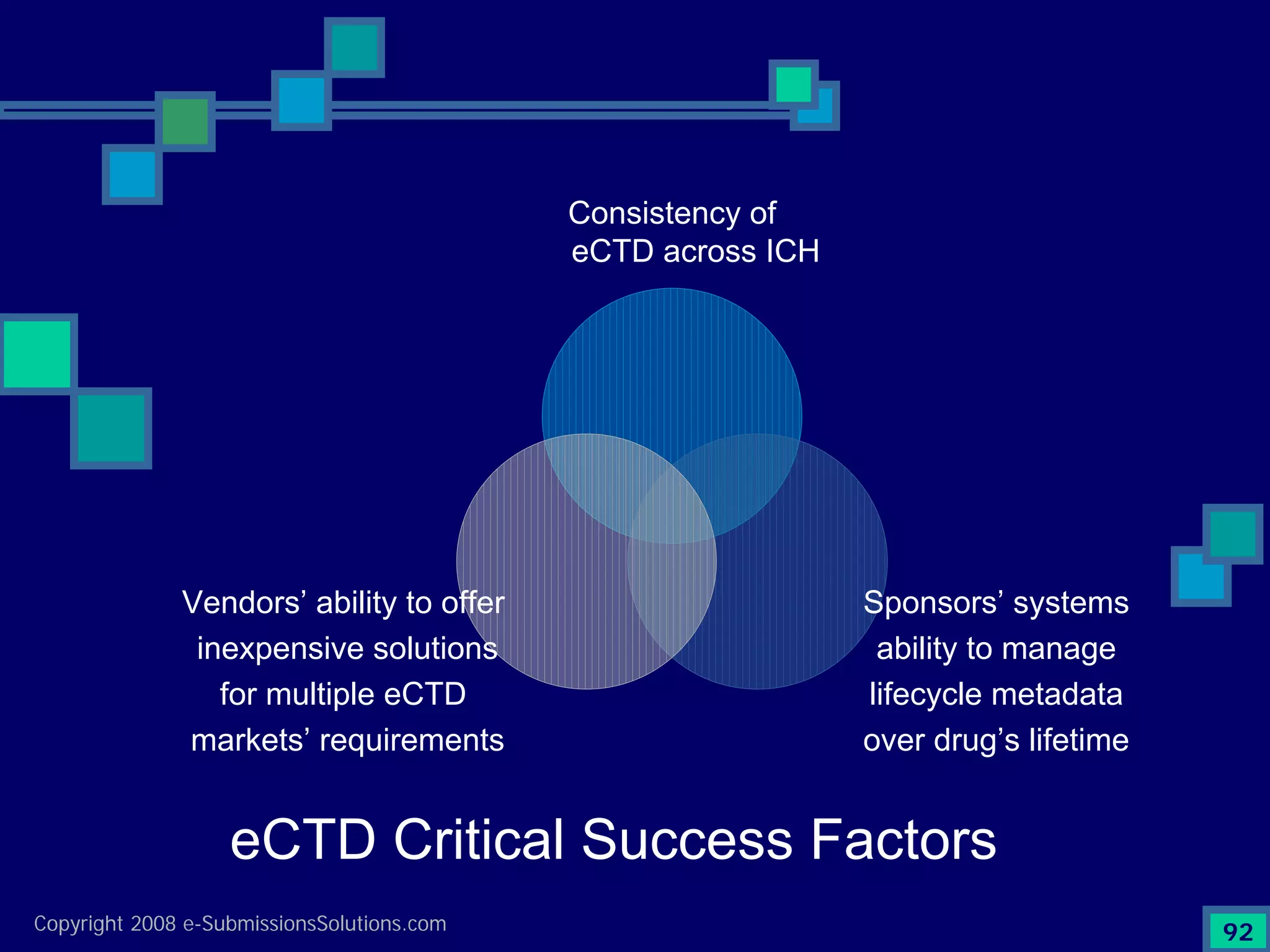
Wraps up the discussion on the critical success factors for eCTD and invites questions for further discussion.



















![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)



































































